Identifying Gene Expression Patterns Associated with Drug-Specific
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins
International Journal of Molecular Sciences Article Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins M. Quadir Siddiqui 1,† , Maulik D. Badmalia 1,† and Trushar R. Patel 1,2,3,* 1 Alberta RNA Research and Training Institute, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive, Lethbridge, AB T1K 3M4, Canada; [email protected] (M.Q.S.); [email protected] (M.D.B.) 2 Department of Microbiology, Immunology and Infectious Disease, Cumming School of Medicine, University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada 3 Li Ka Shing Institute of Virology, University of Alberta, Edmonton, AB T6G 2E1, Canada * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Members of the human Zyxin family are LIM domain-containing proteins that perform critical cellular functions and are indispensable for cellular integrity. Despite their importance, not much is known about their structure, functions, interactions and dynamics. To provide insights into these, we used a set of in-silico tools and databases and analyzed their amino acid sequence, phylogeny, post-translational modifications, structure-dynamics, molecular interactions, and func- tions. Our analysis revealed that zyxin members are ohnologs. Presence of a conserved nuclear export signal composed of LxxLxL/LxxxLxL consensus sequence, as well as a possible nuclear localization signal, suggesting that Zyxin family members may have nuclear and cytoplasmic roles. The molecular modeling and structural analysis indicated that Zyxin family LIM domains share Citation: Siddiqui, M.Q.; Badmalia, similarities with transcriptional regulators and have positively charged electrostatic patches, which M.D.; Patel, T.R. -

Tanibirumab (CUI C3490677) Add to Cart
5/17/2018 NCI Metathesaurus Contains Exact Match Begins With Name Code Property Relationship Source ALL Advanced Search NCIm Version: 201706 Version 2.8 (using LexEVS 6.5) Home | NCIt Hierarchy | Sources | Help Suggest changes to this concept Tanibirumab (CUI C3490677) Add to Cart Table of Contents Terms & Properties Synonym Details Relationships By Source Terms & Properties Concept Unique Identifier (CUI): C3490677 NCI Thesaurus Code: C102877 (see NCI Thesaurus info) Semantic Type: Immunologic Factor Semantic Type: Amino Acid, Peptide, or Protein Semantic Type: Pharmacologic Substance NCIt Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor tyrosine kinase expressed by endothelial cells, while VEGF is overexpressed in many tumors and is correlated to tumor progression. PDQ Definition: A fully human monoclonal antibody targeting the vascular endothelial growth factor receptor 2 (VEGFR2), with potential antiangiogenic activity. Upon administration, tanibirumab specifically binds to VEGFR2, thereby preventing the binding of its ligand VEGF. This may result in the inhibition of tumor angiogenesis and a decrease in tumor nutrient supply. VEGFR2 is a pro-angiogenic growth factor receptor -

LPP (8B3A11) Mouse Mab A
Revision 1 C 0 2 - t LPP (8B3A11) Mouse mAb a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 9 8 Web: [email protected] 3 www.cellsignal.com 3 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source/Isotype: UniProt ID: Entrez-Gene Id: WB, IP, IHC-P, IF-IC H M Hm Mk Endogenous 75 Mouse IgG1 Q93052 4026 Product Usage Information Application Dilution Western Blotting 1:1000 Immunoprecipitation 1:50 Immunohistochemistry (Paraffin) 1:100 Immunofluorescence (Immunocytochemistry) 1:100 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity LPP (8B3A11) Mouse mAb detects endogenous levels of total LPP protein. Species Reactivity: Human, Mouse, Hamster, Monkey Source / Purification Monoclonal antibody is produced by immunizing animals with a HIS-tagged recombinant fragment of human LPP. Background LIM-containing lipoma-preferred partner (LPP) belongs to the zyxin family, members of which include LIMD1, ajuba, trip6 and zyxin. Three LIM domains at the carboxy-terminus characterize this family of proteins. Zyxin family members associate with the actin cytoskeleton and are components of both the cell-cell junction adhesive complex and the integrin-mediated adhesive complex (1). They shuttle in and out of the nucleus where they may function in transcriptional activation (1). LPP binding partners at cell-cell contacts include the actin regulator α-actinin (2) and the human tumor suppressor scrib which regulates cell migration and polarity (3). -

Purging and Haemopoietic Progenitor Cell Selection by CD34 Cell Separation
Bone Marrow Transplantation, (1998) 21, 665–671 1998 Stockton Press All rights reserved 0268–3369/98 $12.00 Purging and haemopoietic progenitor cell selection by CD34؉ cell separation ¨ ¨ W Kruger1, M Gruber1, S Hennings1, N Fehse1, B Fehse1, K Gutensohn2, N Kroger1 and AR Zander1 1Bone Marrow Transplantation Unit, 2Blood Transfusion Service, University Hospital Eppendorf, Hamburg, Germany Summary: High-dose chemotherapy supported by autologous stem cell reinfusion for treatment of metastatic and high-risk female breast cancer is currently under investigation in several Tumour cell contamination of autologous peripheral American and European studies.1,2,3 Tumour cell contami- blood stem cell samples (PBSC) and bone marrow (BM) nation of autologous stem cell products has been described is frequent. Enrichment of CD34+ stem cells is a promis- using immunocytochemistry, reverse transcriptase PCR and ing approach to purging tumour cells from autografts cell culture techniques in approximately one third of har- without damaging progenitor cells. Breast cancer cells vests with a range from 0% to 100%.4–7 The clonogenic were seeded (10؊3؊10؊7) into mononuclear cells from and metastatic potential of tumour cells isolated from blood G-CSF-mobilised PBSC and BM harvests from patients and autografts was demonstrated in cell culture assays and without breast cancer. CD34+ cells were enriched from in nude mice.8–10 The metastatic potential of accidentally mixtures either by immunomagnetic separation (Isolex- retransplanted tumour cells could be investigated by gene 50, and MiniMACS) or by biotin-streptavidin immu- marking of autografts prior to infusion followed by the noaffinity columns (Ceprate-LC). CD34؉ cell fractions detection of marked cells after relapse.11 However, due to were determined by FACS, cancer cells were detected the poor transduction efficacy of epithelial cancer cells by immunocytochemically with an anti-pancytokeratin gene transfer, a negative result would not exclude the possi- antibody. -

The Association of Toll-Like Receptor Polymorphisms With
THE ASSOCIATION OF TOLL-LIKE RECEPTOR POLYMORPHISMS WITH HUMAN IMMUNODEFICIENCY VIRUS INFECTION IN NORTH AMERICANS by BARNE WILLIE Submitted in partial fulfillment of the requirement for the degree of Master of Science Dissertation Advisor: Dr. Peter A Zimmerman (PhD) Biology Case Western Reserve University May 2014 Case Western Reserve University School of Graduate Studies We hereby approve the thesis/dissertation of BARNE WILLIE ………………………………………………………………………………………………………………… candidate for the Master in Science degree*. (signed) Roy Ritzmann (PhD) …………………………………………………………………………………………………………………… (Chair of the committee) Peter A Zimmerman (PhD) …………………………………………………………………………………………………………………… Daniel Tisch (PhD) …………………………………………………………………………………………………………………… Christopher Cullis (PhD) …………………………………………………………………………………………………………………… (date) 21st March 2014 ………………………………………………… * We also certify that the written approval has been obtained for any proprietary material contained therein. II Dedication I would take this precious time that I have to dedicate this work to my Bikpela Papa - Naiyo Niniko and Bikpela Ankol - Phillip Felix Buri. I know you twos will be proud to see me through this time of my life journey. But now, I could only wish you twos were here to witness this. Miss you twos so much, and what more can I say...? I shall see you twos one day. III Table of Content List of Tables ............................................................................................................... VII List of Figures ............................................................................................................ -

Cep-2020-00633.Pdf
Clin Exp Pediatr Vol. 64, No. 5, 208–222, 2021 Review article CEP https://doi.org/10.3345/cep.2020.00633 Understanding the genetics of systemic lupus erythematosus using Bayesian statistics and gene network analysis Seoung Wan Nam, MD, PhD1,*, Kwang Seob Lee, MD2,*, Jae Won Yang, MD, PhD3,*, Younhee Ko, PhD4, Michael Eisenhut, MD, FRCP, FRCPCH, DTM&H5, Keum Hwa Lee, MD, MS6,7,8, Jae Il Shin, MD, PhD6,7,8, Andreas Kronbichler, MD, PhD9 1Department of Rheumatology, Wonju Severance Christian Hospital, Yonsei University Wonju College of Medicine, Wonju, Korea; 2Severance Hospital, Yonsei University College of Medicine, Seoul, Korea; 3Department of Nephrology, Yonsei University Wonju College of Medicine, Wonju, Korea; 4Division of Biomedical Engineering, Hankuk University of Foreign Studies, Yongin, Korea; 5Department of Pediatrics, Luton & Dunstable University Hospital NHS Foundation Trust, Luton, UK; 6Department of Pediatrics, Yonsei University College of Medicine, Seoul, Korea; 7Division of Pediatric Nephrology, Severance Children’s Hospital, Seoul, Korea; 8Institute of Kidney Disease Research, Yonsei University College of Medicine, Seoul, Korea; 9Department of Internal Medicine IV (Nephrology and Hypertension), Medical University Innsbruck, Innsbruck, Austria 1,3) The publication of genetic epidemiology meta-analyses has analyses have redundant duplicate topics and many errors. increased rapidly, but it has been suggested that many of the Although there has been an impressive increase in meta-analyses statistically significant results are false positive. In addition, from China, particularly those on genetic associa tions, most most such meta-analyses have been redundant, duplicate, and claimed candidate gene associations are likely false-positives, erroneous, leading to research waste. In addition, since most suggesting an urgent global need to incorporate genome-wide claimed candidate gene associations were false-positives, cor- data and state-of-the art statistical inferences to avoid a flood of rectly interpreting the published results is important. -

DNA Sequence of the Serratia Marcescens Lipoprotein Gene
Proc. NatI. Acad. Sci. USA Vol. 77, No. 3, pp. 1369-1373, March 1980 Biochemistry DNA sequence of the Serratia marcescens lipoprotein gene (promoter/A-T base pairs/secretion/signal peptide/loop model/gene evolution) KENZO NAKAMURA AND MASAYORI INOUYE Department of Biochemistry, State University of New York, Stony Brook, New York 11794 Communicated by Charles Yanofsky, December 12, 1979 ABSTRACT The Serratia marcescens gene for the outer gene of S. marcescens with that of E. coli revealed several in- membrane lipoprotein (lpp) was cloned in A phage vector teresting and unique features concerning the mechanism of Charon 14. The recombinant phage was very unstable, and the expression of this gene, the secretion of the lipoprotein across Ipp gene with a 300-base-pair deletion at the transcription ter- mination site was further cloned in pBR322. The DNA sequence the cytoplasmic membrane, and the evolution of the gene. of 834 base pairs encompassing the Ipp gene was determined and compared with that of the Escherichia coli ipp gene. The MATERIALS AND METHODS sequence comparisons exhibit several unique features. (i) The Materials. [y-32P]ATP (m9000 Ci/mmol; 1 Ci = 3.7 X 101O promoter region is highly conserved (84% homology) and has becquerels), prepared by the method of Johnson and Walseth an extremely high A+T content (78%) as in E. coli (80%). (ii) (8) from carrier-free The 5' nontranslated region of the lipoprotein mRNA is also [32P]orthophosphate (New England Nu- highly conserved (95% homology). (iii) In the DNA sequence clear) and ADP (Sigma), was used for 5'-end-labeling of DNA corresponding to the signal peptide of this secretory protein, fragments. -

The Chromosome 3P21.3-Encoded Gene, LIMD1, Is a Critical Tumor Suppressor Involved in Human Lung Cancer Development
The chromosome 3p21.3-encoded gene, LIMD1, is a critical tumor suppressor involved in human lung cancer development Tyson V. Sharpa, Ahmad Al-Attarb,1, Daniel E. Foxlera,1, Li Dingc, Thomas Q. de A. Vallima, Yining Zhanga, Hala S. Nijmeha, Thomas M. Webba, Andrew G. Nicholsond, Qunyuan Zhange, Aldi Krajae, Ian Spendloveb, John Osbornec, Elaine Mardisc,e, and Gregory D. Longmoref,2 aSchool of Biomedical Sciences, Queen’s Medical Centre, University of Nottingham Medical School, Nottingham NG7 2UH, United Kingdom; bAcademic and Clinical Departments of Oncology, University of Nottingham, Nottingham NG5 1PB, United Kingdom; dDepartment of Histopathology, Royal Brompton Hospital, London SW3 6NP, United Kingdom; cDepartment of Genetics, Genome Sequencing Center, and eDivision of Statistical Genomics, Washington University School of Medicine, St. Louis, MO 63108; and fDepartments of Medicine and Cell Biology, Washington University, St Louis, MO 63110 Edited by George Klein, Karolinska Institutet, Stockholm, Sweden, and approved October 7, 2008 (received for review May 23, 2008) Loss of heterozygosity (LOH) and homozygous deletions at chro- toma protein (pRB), inhibits E2F-mediated transcription, and mosome 3p21.3 are common in both small and nonsmall cell lung suppresses expression of the majority of genes with E2F1- cancers, indicating the likely presence of tumor suppressor genes responsive elements (9). Moreover, forced expression of LIMD1 (TSGs). Although genetic and epigenetic changes within this region in A549 lung tumor cell line blocks tumor growth in vitro and in have been identified, the functional significance of these changes vivo (9). Here, we show that expression of the 3p21.3 gene, has not been explored. -

Fine Mapping of the Celiac Disease-Associated LPP Locus Reveals a Potential Functional Variant
Human Molecular Genetics, 2014, Vol. 23, No. 9 2481–2489 doi:10.1093/hmg/ddt619 Advance Access published on December 11, 2013 Fine mapping of the celiac disease-associated LPP locus reveals a potential functional variant Rodrigo Almeida1,2,{, Isis Rican˜ o-Ponce1,{, Vinod Kumar1, Patrick Deelen1, Agata Szperl1, Gosia Trynka1,{, Javier Gutierrez-Achury1, Alexandros Kanterakis1, Harm-Jan Westra1, Lude Franke1, Morris A. Swertz1, Mathieu Platteel1, Jose Ramon Bilbao3, Donatella Barisani4, Luigi Greco5, Luisa Mearin6, Victorien M. Wolters7, Chris Mulder8, Maria Cristina Mazzilli9, Ajit Sood10, Bozena Cukrowska11, Concepcio´ nNu´ n˜ ez12, Riccardo Pratesi2, Sebo Withoff1 Downloaded from https://academic.oup.com/hmg/article/23/9/2481/631913 by guest on 25 September 2021 and Cisca Wijmenga1,∗ 1Department of Genetics, University of Groningen, University Medical Center Groningen, PO Box 30001, Groningen 9700 RB, The Netherlands, 2Graduate Program in Health Sciences, University of Brasilia School of Health Sciences, Brasilia, Brazil, 3Immunogenetics Research Laboratory, Hospital Universitario de Cruces, Barakaldo, Bizkaia 48903, Spain, 4Department of Experimental Medicine, Faculty of Medicine, University of Milano-Bicocca, Monza, Italy, 5European Laboratory for Food Induced Disease, University of Naples Federico II, Naples, Italy, 6Department of Pediatric Gastroenterology, Leiden University Medical Centre, Leiden, The Netherlands, 7Department of Pediatric Gastroenterology, University Medical Centre Utrecht, Utrecht, The Netherlands, 8Department -

A Causal Gene Network with Genetic Variations Incorporating Biological Knowledge and Latent Variables
A CAUSAL GENE NETWORK WITH GENETIC VARIATIONS INCORPORATING BIOLOGICAL KNOWLEDGE AND LATENT VARIABLES By Jee Young Moon A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Statistics) at the UNIVERSITY OF WISCONSIN–MADISON 2013 Date of final oral examination: 12/21/2012 The dissertation is approved by the following members of the Final Oral Committee: Brian S. Yandell. Professor, Statistics, Horticulture Alan D. Attie. Professor, Biochemistry Karl W. Broman. Professor, Biostatistics and Medical Informatics Christina Kendziorski. Associate Professor, Biostatistics and Medical Informatics Sushmita Roy. Assistant Professor, Biostatistics and Medical Informatics, Computer Science, Systems Biology in Wisconsin Institute of Discovery (WID) i To my parents and brother, ii ACKNOWLEDGMENTS I greatly appreciate my adviser, Prof. Brian S. Yandell, who has always encouraged, inspired and supported me. I am grateful to him for introducing me to the exciting research areas of statis- tical genetics and causal gene network analysis. He also allowed me to explore various statistical and biological problems on my own and guided me to see the problems in a bigger picture. Most importantly, he waited patiently as I progressed at my own pace. I would also like to thank Dr. Elias Chaibub Neto and Prof. Xinwei Deng who my adviser arranged for me to work together. These three improved my rigorous writing and thinking a lot when we prepared the second chapter of this dissertation for publication. It was such a nice opportunity for me to join the group of Prof. Alan D. Attie, Dr. Mark P. Keller, Prof. Karl W. Broman and Prof. -
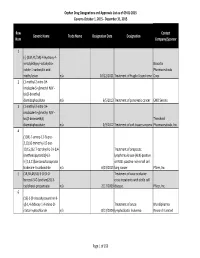
Orphan Drug Designation List
Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 1 (‐)‐(3aR,4S,7aR)‐4‐Hydroxy‐4‐ m‐tolylethynyl‐octahydro‐ Novartis indole‐1‐carboxylic acid Pharmaceuticals methyl ester n/a 10/12/2011 Treatment of Fragile X syndrome Corp. 2 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐broethyl) diamidophosphate n/a 6/5/2013 Treatment of pancreatic cancer EMD Serono 3 (1‐methyl‐2‐nitro‐1H‐ imidazole‐5‐yl)methyl N,N'‐ bis(2‐bromoethyl) Threshold diamidophosphate n/a 3/9/2012 Treatment of soft tissue sarcoma Pharmaceuticals, Inc. 4 (1OR)‐7‐amino‐12‐fluoro‐ 2,10,16‐trimethyl‐15 oxo‐ 10,15,16,17‐tetrahydro‐2H‐8,4‐ Treatment of anaplastic (metheno)pyrazolo[4,3‐ lymphoma kinase (ALK)‐positive h][2,5,11]benzoxadiazacyclote or ROS1‐positive non‐small cell tradecine‐3‐carbonitrile n/a 6/23/2015 lung cancer Pfizer, Inc. 5 (1R,3R,4R,5S)‐3‐O‐[2‐O‐ Treatment of vaso‐occlusive benzoyl‐3‐O‐(sodium(2S)‐3‐ crisis in patients with sickle cell cyclohexyl‐propanoate‐ n/a 2/17/2009 disease. Pfizer, Inc. 6 (1S)‐1‐(9‐deazahypoxanthin‐9‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ Treatment of acute Mundipharma ribitol‐hydrochloride n/a 8/13/2004 lymphoblastic leukemia Research Limited Page 1 of 359 Orphan Drug Designations and Approvals List as of 09‐01‐2015 Governs October 1, 2015 ‐ December 31, 2015 Row Contact Generic Name Trade Name Designation Date Designation Num Company/Sponsor 7 Treatment of chronic lymphocytic leukemia and related leukemias to include (1S)‐1‐(9‐deazahypoxanthin‐9‐ prolymphocytic leukemia, adult T‐ yl)‐1,4‐dideoxy‐1,4‐imino‐D‐ cell leukemia, and hairy cell Mundipharma ribitol‐hydrochloride n/a 8/10/2004 leukemia Research Ltd. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0226431 A1 Habib (43) Pub
US 20090226431A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0226431 A1 Habib (43) Pub. Date: Sep. 10, 2009 (54) TREATMENT OF CANCER AND OTHER Publication Classification DISEASES (51) Int. Cl. A 6LX 3/575 (2006.01) (76)76) InventorInventor: Nabilabil Habib,Habib. Beirut (LB(LB) C07J 9/00 (2006.01) Correspondence Address: A 6LX 39/395 (2006.01) 101 FEDERAL STREET A6IP 29/00 (2006.01) A6IP35/00 (2006.01) (21) Appl. No.: 12/085,892 A6IP37/00 (2006.01) 1-1. (52) U.S. Cl. ...................... 424/133.1:552/551; 514/182: (22) PCT Filed: Nov.30, 2006 514/171 (86). PCT No.: PCT/US2O06/045665 (57) ABSTRACT .."St. Mar. 6, 2009 The present invention relates to a novel compound (e.g., 24-ethyl-cholestane-3B.5C,6C.-triol), its production, its use, and to methods of treating neoplasms and other tumors as Related U.S. Application Data well as other diseases including hypercholesterolemia, (60) Provisional application No. 60/741,725, filed on Dec. autoimmune diseases, viral diseases (e.g., hepatitis B, hepa 2, 2005. titis C, or HIV), and diabetes. F2: . - 2 . : F2z "..., . Cz: ".. .. 2. , tie - . 2 2. , "Sphagoshgelin , , re Cls Phosphatidiglethanolamine * - 2 .- . t - r y ... CBs .. A . - . Patent Application Publication Sep. 10, 2009 Sheet 1 of 16 US 2009/0226431 A1 E. e'' . Phosphatidylcholine. " . Ez'.. C.2 . Phosphatidylserias. * . - A. z' C. w E. a...2 .". is 2 - - " - B 2. Sphingoshgelin . Cls Phosphatidglethanglamine Figure 1 Patent Application Publication Sep. 10, 2009 Sheet 2 of 16 US 2009/0226431 A1 Chile Phosphater Glycerol Phosphatidylcholine E.