Moths of North Carolina - Early Draft 1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Checklist of Texas Lepidoptera Knudson & Bordelon, Jan 2018 Texas Lepidoptera Survey
1 Checklist of Texas Lepidoptera Knudson & Bordelon, Jan 2018 Texas Lepidoptera Survey ERIOCRANIOIDEA TISCHERIOIDEA ERIOCRANIIDAE TISCHERIIDAE Dyseriocrania griseocapitella (Wlsm.) Eriocraniella mediabulla Davis Coptotriche citripennella (Clem.) Eriocraniella platyptera Davis Coptotriche concolor (Zell.) Coptotriche purinosella (Cham.) Coptotriche clemensella (Cham). Coptotriche sulphurea (F&B) NEPTICULOIDEA Coptotriche zelleriella (Clem.) Tischeria quercitella Clem. NEPTICULIDAE Coptotriche malifoliella (Clem.) Coptotriche crataegifoliae (Braun) Ectoedemia platanella (Clem.) Coptotriche roseticola (F&B) Ectoedemia rubifoliella (Clem.) Coptotriche aenea (F&B) Ectoedemia ulmella (Braun) Asterotriche solidaginifoliella (Clem.) Ectoedemia obrutella (Zell.) Asterotriche heliopsisella (Cham.) Ectoedemia grandisella (Cham.) Asterotriche ambrosiaeella (Cham.) Nepticula macrocarpae Free. Asterotriche helianthi (F&B) Stigmella scintillans (Braun) Asterotriche heteroterae (F&B) Stigmella rhoifoliella (Braun) Asterotriche longeciliata (F&B) Stigmella rhamnicola (Braun) Asterotriche omissa (Braun) Stigmella villosella (Clem.) Asterotriche pulvella (Cham.) Stigmella apicialbella (Cham.) Stigmella populetorum (F&B) Stigmella saginella (Clem.) INCURVARIOIDEA Stigmella nigriverticella (Cham.) Stigmella flavipedella (Braun) PRODOXIDAE Stigmella ostryaefoliella (Clem.) Stigmella myricafoliella (Busck) Tegeticula yuccasella (Riley) Stigmella juglandifoliella (Clem.) Tegeticula baccatella Pellmyr Stigmella unifasciella (Cham.) Tegeticula carnerosanella Pellmyr -

Molecular Phylogeny and Systematics of the Pieridae (Lepidoptera: Papilionoidea): Higher Classification and Biogeography
Blackwell Publishing LtdOxford, UKZOJZoological Journal of the Linnean Society0024-4082The Lin- nean Society of London, 2006? 2006 147? 239275 Original Article PHYLOGENY AND SYSTEMATICS OF THE PIERIDAEM. F. BRABY ET AL. Zoological Journal of the Linnean Society, 2006, 147, 239–275. With 8 figures Molecular phylogeny and systematics of the Pieridae (Lepidoptera: Papilionoidea): higher classification and Downloaded from https://academic.oup.com/zoolinnean/article-abstract/147/2/239/2631026 by Harvard Library user on 21 November 2018 biogeography MICHAEL F. BRABY1,2*, ROGER VILA1 and NAOMI E. PIERCE1 1Museum of Comparative Zoology, Harvard University, 26 Oxford St, Cambridge, MA 02138, USA 2School of Botany and Zoology, The Australian National University, Canberra, ACT 0200, Australia Received May 2004; accepted for publication October 2005 The systematic relationships of the butterfly family Pieridae are poorly understood. Much of our current under- standing is based primarily on detailed morphological observations made 50–70 years ago. However, the family and its putative four subfamilies and two tribes, have rarely been subjected to rigorous phylogenetic analysis. Here we present results based on an analysis of molecular characters used to reconstruct the phylogeny of the Pieridae in order to infer higher-level classification above the generic level and patterns of historical biogeography. Our sample contained 90 taxa representing 74 genera and six subgenera, or 89% of all genera recognized in the family. Three complementary approaches were -

Peter Buck Fellowship Program 2014 - 2015 Progress Report
The Peter Buck Fellowship Program 2014 - 2015 Progress Report Overview This year, the National Museum of Natural History’s Peter Buck Fellowship Program welcomes its fifth class of participants. Reaching this milestone offers a unique perspective, both for reflecting on the Buck Program’s past success and for envisioning the course of its future. Through contributions to research, publications, and educational outreach across all of the Museum’s scientific departments, each class further defines the scope and impact of what it means to be a Peter Buck Fellow. Throughout the Smithsonian and scientific communities, their work is advancing learning about nature and culture—knowledge that ultimately drives solutions for the global challenges we face. For the 67 predoctoral and postdoctoral fellows admitted since the Program began, the experience is a pivotal step in advancing their science careers. The Peter Buck Fellowship Program currently includes a robust group of 34 fellows in residence or anticipated in the next few months. The most recent 14 awardees, selected as the Class of 2015, hail from a variety of academic institutions throughout the United States and beyond. A growing alumni base—currently made up of 27 individuals who have since completed their tenure at the Museum—provides yet another lens for understanding the far-reaching effects of the fellowship experience as they pursue the next phase of their careers. This year also welcomes the first recipients of Buck Fellowships contributing specifically to the goals of two major Museum initiatives. Deep Time fellowship opportunities are aimed at increasing the research, education, and outreach productivity of the Initiative during the period leading up to the opening of the permanent Fossil Hall in 2019. -

THE MICROLEPIDOPTERA Section 2 COSMOPTERIGIDAE THROUGH HEPIALIDAE
The University of Maine DigitalCommons@UMaine Technical Bulletins Maine Agricultural and Forest Experiment Station 8-1-1984 TB114: A List of the Lepidoptera of Maine--Part 2: The icrM olepidoptera Section 2 Cosmopterigidae through Hepialidae Auburn E. Brower Follow this and additional works at: https://digitalcommons.library.umaine.edu/aes_techbulletin Part of the Entomology Commons Recommended Citation Brower, A.E. 1984. A list of the Lepidoptera of Maine--Part 2: The icrM olepidoptera section 2 Cosmopterigidae through Hepialidae. Maine Agricultural Experiment Station Technical Bulletin 114. This Article is brought to you for free and open access by DigitalCommons@UMaine. It has been accepted for inclusion in Technical Bulletins by an authorized administrator of DigitalCommons@UMaine. For more information, please contact [email protected]. ISSN 0734-9556 A LIST OF THE LEPIDOPTERA OF MAINE Part 2 THE MICROLEPIDOPTERA Section 2 COSMOPTERIGIDAE THROUGH HEPIALIDAE Auburn E. Brower A GHOST MOTH — Sthenopis argenteomaculatus Harris A JOINT PUBLICATION OF THE (MAINE DEPARTMENT OF CONSERVATION Maine Forest Service Division of Entomology, Augusta, Maine and the DEPARTMENT OF ENTOMOLOGY, ORONO August 1984 Inquiries concerning this bulletin may be sent to: Dr. Auburn E. Brower 8 Hospital Street Augusta, ME A LIST OF THE LEPIDOPTERA OF MAINE Part 2 THE MICROLEPIDOPTERA Section 2 COSMOPTERIGIDAE THROUGH HEPIALIDAE Auburn E. Brower A JOINT PUBLICATION OF THE MAINE DEPARTMENT OF CONSERVATION Maine Forest Service Division of Entomology, -

A Molecular Phylogeny for Yponomeutoidea (Insecta, Lepidoptera, Ditrysia) and Its Implications for Classification, Biogeography and the Evolution of Host Plant Use
A Molecular Phylogeny for Yponomeutoidea (Insecta, Lepidoptera, Ditrysia) and Its Implications for Classification, Biogeography and the Evolution of Host Plant Use Jae-Cheon Sohn1*, Jerome C. Regier1, Charles Mitter1, Donald Davis2, Jean-Franc¸ois Landry3, Andreas Zwick4, Michael P. Cummings5 1 Department of Entomology, University of Maryland, College Park, Maryland, United States of America, 2 Department of Entomology, National Museum of Natural History, Smithsonian Institution, Washington DC, United States of America, 3 Agriculture and Agri-Food Canada, Eastern Cereal and Oilseed Research Centre, C.E.F., Ottawa, Canada, 4 Department of Entomology, State Museum of Natural History, Stuttgart, Germany, 5 Laboratory of Molecular Evolution, Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland, United States of America Abstract Background: Yponomeutoidea, one of the early-diverging lineages of ditrysian Lepidoptera, comprise about 1,800 species worldwide, including notable pests and insect-plant interaction models. Yponomeutoids were one of the earliest lepidopteran clades to evolve external feeding and to extensively colonize herbaceous angiosperms. Despite the group’s economic importance, and its value for tracing early lepidopteran evolution, the biodiversity and phylogeny of Yponomeutoidea have been relatively little studied. Methodology/Principal Findings: Eight nuclear genes (8 kb) were initially sequenced for 86 putative yponomeutoid species, spanning all previously recognized suprageneric groups, and 53 outgroups representing 22 families and 12 superfamilies. Eleven to 19 additional genes, yielding a total of 14.8 to 18.9 kb, were then sampled for a subset of taxa, including 28 yponomeutoids and 43 outgroups. Maximum likelihood analyses were conducted on data sets differing in numbers of genes, matrix completeness, inclusion/weighting of synonymous substitutions, and inclusion/exclusion of ‘‘rogue’’ taxa. -

Search: Thu Nov 5 14:40:21 2020Page 1 Of
Search: Thu Nov 5 14:40:21 2020 Page 1 of 180 10 % Athalia cornubiae|[1]|GBSYM1130-12|Hymenoptera|BOLD:AAJ9512 Sciaridae|[2]|CNFNR1642-14|Diptera|BOLD:ACM8049 Sciaridae|[3]|CNPPB684-12|Diptera|BOLD:ACC8493 Sciaridae|[4]|GMGSL144-13|Diptera|BOLD:ACC8327 Sciaridae|[5]|GMOJG309-15|Diptera|BOLD:ACX6391 Tetraneura nigriabdominalis|[6]|ASHMT220-11|Hemiptera|BOLD:AAG3896 Tetraneura ulmi|[7]|CNJAE749-12|Hemiptera|BOLD:AAG3894 Prociphilus caryae|[8]|PHOCT611-11|Hemiptera|BOLD:ABY5255 Prociphilus tessellatus|[9]|TTSOW205-10|Hemiptera|BOLD:AAD7311 Grylloprociphilus imbricator|[10]|RDBA378-06|Hemiptera|BOLD:AAY2624 Subsaltusaphis virginica|[11]|RFBAC272-07|Hemiptera|BOLD:AAH9929 Euceraphis papyrifericola|[12]|BBHCN315-10|Hemiptera|BOLD:AAH2870 Euceraphis|[13]|CNPAI428-13|Hemiptera|BOLD:AAX7972 Calaphis betulaecolens|[14]|ASAHE150-12|Hemiptera|BOLD:AAC3672 Callipterinella calliptera|[15]|RDBA165-05|Hemiptera|BOLD:AAB6094 Periphyllus negundinis|[16]|CNEIB825-12|Hemiptera|BOLD:AAD3938 Sipha elegans|[17]|CNEIF3636-12|Hemiptera|BOLD:AAG1528 Aphididae|[18]|GMGSW027-13|Hemiptera|BOLD:ACD2286 Drepanaphis|[19]|RRMFE3184-15|Hemiptera|BOLD:ABY0945 Drepanaphis|[20]|RDBA009-05|Hemiptera|BOLD:AAH2879 Drepanaphis|[21]|BBHEM601-10|Hemiptera|BOLD:AAX8898 Drepanaphis|[22]|GMGSA077-12|Hemiptera|BOLD:ACA3956 Drepanaphis|[23]|ASAHE012-12|Hemiptera|BOLD:ABY1338 Drepanaphis|[24]|RRSSA5353-15|Hemiptera|BOLD:AAI6141 Drepanaphis parva|[25]|CNSLF196-12|Hemiptera|BOLD:AAX8895 Drepanaphis|[26]|CNPEE1905-14|Hemiptera|BOLD:ACL5395 Drepanaphis acerifoliae|[27]|RFBAG183-11|Hemiptera|BOLD:AAH2868 -

Lepidoptera: Pieridae
Blackwell Publishing LtdOxford, UKBIJBiological Journal of the Linnean Society0024-4066© 2007 The Linnean Society of London? 2007 90? 413440 Original Article PHYLOGENY AND BIOGEOGRAPHY OF THE APORIINA M. F. BRABY ET AL. Biological Journal of the Linnean Society, 2007, 90, 413–440. With 7 figures Phylogeny and historical biogeography of the subtribe Aporiina (Lepidoptera: Pieridae): implications for the Downloaded from https://academic.oup.com/biolinnean/article-abstract/90/3/413/2701092 by Harvard University user on 21 November 2018 origin of Australian butterflies MICHAEL F. BRABY1,2*, NAOMI E. PIERCE1 and ROGER VILA1† 1Department of Organismic and Evolutionary Biology, Harvard University, 26 Oxford Street, Cambridge, MS 02138, USA 2School of Botany and Zoology, The Australian National University, Canberra, ACT 0200, Australia Received 13 July 2005; accepted for publication 1 May 2006 The Australian fauna is composed of several major biogeographical elements reflecting different spatial and tem- poral histories. Two groups of particular interest are the Gondwanan Element, reflecting an ancient origin in Gond- wana or southern Gondwana (southern vicariance hypothesis), and the Asian Element, reflecting a more recent origin in Asia, Eurasia or Laurasia (northern dispersal hypothesis). Theories regarding the origin and evolution of butterflies (Hesperioidea, Papilionoidea) in Australia are controversial, with no clear consensus. Here, we investigate the phylogenetic and historical biogeographical relationships of the subtribe Aporiina, a widespread taxon with dis- junct distributions in each of the major zoogeographical regions. Attention is paid to origins of the subtribe in the Australian Region for which several conflicting hypotheses have been proposed for the Old World genus Delias Hüb- ner. -
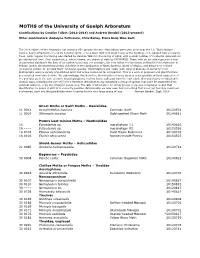
Moth Records
MOTHS of the University of Guelph Arboretum Contributions by Candice Talbot (2012-2014) and Andrew Bendall (2013-present) Other contributors: Andalyne Tofflemire, Chris Earley, Fiona Reid, Mike Kent The 2018 edition of the Arboretum list contains 851 species of moth. Most adults were seen at or near the J.C. Taylor Nature Centre, being attracted to the white building lights, or to a black light and sheet hung by the building, or to painted bait on nearby trees. Semi-regular monitoring was started by Candice Talbot in the spring of 2012, with a small number of incidental observations pre-dating that time. First record dates, where known, are shown at right as YYYYMMDD. Those with an asterisk represent a first documented sighting if the date of an earlier record was not available. We now follow the taxonomy outlined in Pohl, Patterson & Pelham (2016) Annotated taxonomic checklist of the Lepidoptera of North America, North of Mexico, and adopt their revised numbering system for all valid North American species. Identifications are made (with varying degrees of certainty) from photographs using a variety of published print and online resources for comparison. This is a work in progress and identifications are revisited from time to time. We acknowledge that definitive identification of some species is not possible without dissection of the genitalia or, in the case of some microlepidoptera, rearing larvae collected from the host plant. Our uncertainty is indicated in various ways, including the use of [t] for a tentative identification, by indicating a group of species that can't be separated from external features, or by identifying to genus only. -

Predator to Prey to Poop: Bats As Microbial Hosts and Insectivorous Hunters
Predator to Prey to Poop: Bats as Microbial Hosts and Insectivorous Hunters A Thesis SUBMITTED TO THE FACULTY OF THE UNIVERSITY OF MINNESOTA BY Miranda Galey IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE Dr. Ron Moen, Dr. Jessica R. Sieber September 2020 Copyright © Miranda Galey 2020 Abstract Bat fecal samples are a rich source of ecological data for bat biologists, entomologists, and microbiologists. Feces collected from individual bats can be used to profile the gut microbiome using microbial DNA and to understand bat foraging strategies using arthropod DNA. We used eDNA collected from bat fecal samples to better understand bats as predators in the context of their unique gut physiology. We used high through- put sequencing of the COI gene and 16S rRNA gene to determine the diet composition and gut microbiome composition of three bat species in Minnesota: Eptesicus fuscus, Myotis lucifugus and M. septentrionalis. In our analysis of insect prey, we found that E. fuscus consistently foraged for a higher diversity of beetle species compared to other insects. We found that the proportional frequency of tympanate samples from M. septentrionalis and M. lucifugus was similar, while M. septentrionalis consistently preyed more often upon non-flying species. We used the same set of COI sequences to determine presence of pest species, rare species, and insects not previously observed in Minnesota. We were able to combine precise arthropod identification and the for- aging areas of individually sampled bats to observe possible range expansion of some insects. The taxonomic composition of the bat gut microbiome in all three species was found to be consistent with the composition of a mammalian small intestine. -

Insect Biodiversity on Prairies Lacking Fire Management
1 Insect Biodiversity on Prairies Lacking Fire Management with focus on Lepidoptera (Moths and Butterflies) Kyle Johnson Honorary Fellow, University of Wisconsin-Madison 445 Russell Labs, 1630 Linden Drive, Madison, WI, 53706 [email protected] 2 Table of Contents Acknowledgements....................................................................................................................................3 Introduction and Study Overview..............................................................................................................3 Study Sites..................................................................................................................................................4 Methods....................................................................................................................................................10 Results.......................................................................................................................................................14 Discussion..................................................................................................................................................47 Future Studies............................................................................................................................................48 References.................................................................................................................................................48 3 Acknowledgements This project was financially supported -
Moths & Butterflies
Moths & Butterflies of Eagle Valley Preserve Compiled by Kyle E. Johnson Data Sources and Acknowledgements This checklist is based almost entirely on specimens collected and determined by the author (470+ species). Andrew Williams contributed seven additional species. All species are based on voucher specimens; these are housed in the University of Wisconsin-Madison Insect Research Collection (WIRC), with a few duplicates in other research collections. The specimens have detailed data and are fully databased. The Kohler Trust for Preservation provided funding for this project. I thank Brett & Carole Mandernack for their hospitality and enthusiasm while visiting the preserve. I thank Dan Young and the University of Wisconsin-Madison Insect Research Collection for the lab space and other resources which make such projects possible. Finally, I thank Bob Borth and Steve Bransky for good company in the field. Micro Moths = 156+ species Butterflies = 35 species Macro Moths = 286+ species Total Lepidoptera = 477+ species Biodiversity on the Preserve How many more species are out there? A whole lot is the short answer. Sampling on the preserve has only spanned two years; there are plenty of weaknesses in seasonal coverage, habitats which have been sparsely investigated, and sampling techniques which need attention. Additional species even lurk among the specimens already collected as some remain unidentified, the vast majority of these being “micro moths”. Thus the micro moths are underrepresented in this checklist. A conservative estimate suggests that 470 species is roughly one third of the actual diversity. At Mosquito Hill Nature Center in Outagamie County Hugo Kons Jr. (pers. comm.) surveyed for macro moths and butterflies intensively over many years; he documented an impressive 782 species. -

Moth Community from a Northcentral Florida LOCATION - a TAXONOMIC CHECKLIST
WARREN ET AL.: Phocides behavior AUSTIN: List of northcentral Florida moths TROP. LEPID. RES., 20(1):41-44, 2010 41 SCIENTIFIC NOTE: MOTH COMMUNITY FROM A northcentral FLORIDA LOCATION - A TAXONOMIC CHECKLIST George T. Austin* McGuire Center for Lepidoptera and Biodiversity, Florida Museum of Natural History, University of Florida, Gainesville, FL MOTHS RECORDED AT 2004 SE 41ST AVENUE, GAINESVILLE, ALACHUA COUNTY, FLORIDA (29º36.95’N 82º17.91’W) BETWEEN 2005 AND 2009 NEPTICULIDAE GRACILLARIIDAE 2209 Stegasta bosqueella 1474 Cosmopterix attenuatella 76+ Stigmella sp. 592+ Caloptilia sp. #1 2211 Polyhymno luteostrigella 1479 Cosmopterix dapifera 96 Stigmella nr. unifasciata 592+ Caloptilia sp. #2 2216 Ymeldia janae 1487 Cosmopterix abdita 592+ Caloptilia sp. #3 2223 Untomia nr. albistrigella 1494 Cosmopterix scirpicola OPOSTEGIDAE 596 Caloptilia nr. blandella 2227+ Battaristis sp. 1495 Cosmopterix ebriola 121 Pseudopostega albogaleriella 620 Caloptilia nr. packardella 2229 Battaristis vittella 1503 Melanocinclis lineigera 630 Caloptilia rhoifoliella 2233 Anacampsis conclusella 1512 Pyroderces rileyi TISCHERIIDAE 641 Caloptilia superbifrontella 2234 Anacampsis coverdalella 1513 Pyroderces badia 130+ Tischeria sp. 644 Caloptilia violacella 2265 Helcystogramma chambersella 1515 Limnaecia phragmitella 130+ Tischeria sp. 663+ Neurobathra sp. 2281 Dichomeris ligulella 1589 Siskiwitia falcata 144 Tischeria nr. quercitella 663 Neurobathra strigifinitella 2284 Dichomeris acuminata 1623 Perimede erransella 700 Lithocolletis sp.? 2284.1 Dichomeris