Hydrogen Elimination for Permethylscandocene Alkyl
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catalytic Pyrolysis of Plastic Wastes for the Production of Liquid Fuels for Engines
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supporting information for: Catalytic pyrolysis of plastic wastes for the production of liquid fuels for engines Supattra Budsaereechaia, Andrew J. Huntb and Yuvarat Ngernyen*a aDepartment of Chemical Engineering, Faculty of Engineering, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail:[email protected] bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand Fig. S1 The process for pelletization of catalyst PS PS+bentonite PP ) t e PP+bentonite s f f o % ( LDPE e c n a t t LDPE+bentonite s i m s n HDPE a r T HDPE+bentonite Gasohol 91 Diesel 4000 3500 3000 2500 2000 1500 1000 500 Wavenumber (cm-1) Fig. S2 FTIR spectra of oil from pyrolysis of plastic waste type. Table S1 Compounds in oils (%Area) from the pyrolysis of plastic wastes as detected by GCMS analysis PS PP LDPE HDPE Gasohol 91 Diesel Compound NC C Compound NC C Compound NC C Compound NC C 1- 0 0.15 Pentane 1.13 1.29 n-Hexane 0.71 0.73 n-Hexane 0.65 0.64 Butane, 2- Octane : 0.32 Tetradecene methyl- : 2.60 Toluene 7.93 7.56 Cyclohexane 2.28 2.51 1-Hexene 1.05 1.10 1-Hexene 1.15 1.16 Pentane : 1.95 Nonane : 0.83 Ethylbenzen 15.07 11.29 Heptane, 4- 1.81 1.68 Heptane 1.26 1.35 Heptane 1.22 1.23 Butane, 2,2- Decane : 1.34 e methyl- dimethyl- : 0.47 1-Tridecene 0 0.14 2,2-Dimethyl- 0.63 0 1-Heptene 1.37 1.46 1-Heptene 1.32 1.35 Pentane, -

Rare Earth Pincer Complexes: Synthesis, Reaction Chemistry, and Catalysis
Top Organomet Chem (2016) 54: 93–178 DOI: 10.1007/3418_2015_120 # Springer International Publishing Switzerland 2015 Published online: 21 June 2015 Rare Earth Pincer Complexes: Synthesis, Reaction Chemistry, and Catalysis Mikko M. Ha¨nninen, Matthew T. Zamora, and Paul G. Hayes Abstract The research field surrounding rare earth pincer complexes has reached a stage where a comprehensive review about the reactivity and catalytic behavior of these species is justified. In this contribution, we begin with a brief introduction on common strategies for the preparation of rare earth pincer complexes, continuing with a section devoted to the versatile reactivity observed for this class of com- pound. Thereafter, several types of compounds are discussed, including extremely reactive hydrides, cationic species, and intriguing scandium imido complexes. Finally, the last portion of this chapter sums up the hitherto reported catalytic studies, including discussions on ring-opening polymerization of cyclic esters, polymerization of olefins and hydroamination reactions, as well as several exam- ples of more infrequently encountered catalytic processes. Keywords Catalysis Á Lanthanides Á Pincer ligands Á Rare earth metals Á Reactivity Contents 1 Introduction .................................................................................. 94 2 Preparation of Rare Earth Complexes Supported by Pincer Ligands ..................... 95 2.1 Common Synthetic Routes to Rare Earth Pincer Complexes ....................... 95 2.2 Reactions of Metal Complexes That -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”
chemengineering Article Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy” Robert J. Meier Pro-Deo Consultant, 52525 Heinsberg, North-Rhine Westphalia, Germany; [email protected] Abstract: Group contribution (GC) methods to predict thermochemical properties are of eminent importance to process design. Compared to previous works, we present an improved group contri- bution parametrization for the heat of formation of organic molecules exhibiting chemical accuracy, i.e., a maximum 1 kcal/mol (4.2 kJ/mol) difference between the experiment and model, while, at the same time, minimizing the number of parameters. The latter is extremely important as too many parameters lead to overfitting and, therewith, to more or less serious incorrect predictions for molecules that were not within the data set used for parametrization. Moreover, it was found to be important to explicitly account for common chemical knowledge, e.g., geminal effects or ring strain. The group-related parameters were determined step-wise: first, alkanes only, and then only one additional group in the next class of molecules. This ensures unique and optimal parameter values for each chemical group. All data will be made available, enabling other researchers to extend the set to other classes of molecules. Keywords: enthalpy of formation; thermodynamics; molecular modeling; group contribution method; quantum mechanical method; chemical accuracy; process design Citation: Meier, R.J. Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”. 1. Introduction ChemEngineering 2021, 5, 24. To understand chemical reactivity and/or chemical equilibria, knowledge of thermo- o https://doi.org/10.3390/ dynamic properties such as gas-phase standard enthalpy of formation DfH gas is a necessity. -

Measurements of Higher Alkanes Using NO Chemical Ionization in PTR-Tof-MS
Atmos. Chem. Phys., 20, 14123–14138, 2020 https://doi.org/10.5194/acp-20-14123-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Measurements of higher alkanes using NOC chemical ionization in PTR-ToF-MS: important contributions of higher alkanes to secondary organic aerosols in China Chaomin Wang1,2, Bin Yuan1,2, Caihong Wu1,2, Sihang Wang1,2, Jipeng Qi1,2, Baolin Wang3, Zelong Wang1,2, Weiwei Hu4, Wei Chen4, Chenshuo Ye5, Wenjie Wang5, Yele Sun6, Chen Wang3, Shan Huang1,2, Wei Song4, Xinming Wang4, Suxia Yang1,2, Shenyang Zhang1,2, Wanyun Xu7, Nan Ma1,2, Zhanyi Zhang1,2, Bin Jiang1,2, Hang Su8, Yafang Cheng8, Xuemei Wang1,2, and Min Shao1,2 1Institute for Environmental and Climate Research, Jinan University, 511443 Guangzhou, China 2Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, 511443 Guangzhou, China 3School of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), 250353 Jinan, China 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Protection and Resources Utilization, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, 510640 Guangzhou, China 5State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, 100871 Beijing, China 6State Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese -
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
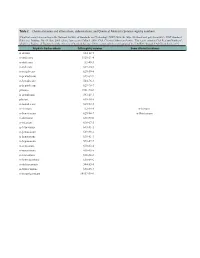
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Thermodynamic Modelling of Hydrocarbon-Chains and Light-Weight Supercritical Solvents
Thermodynamic modelling of hydrocarbon-chains and light-weight supercritical solvents by James Edward Lombard Thesis presented in partial fulfilment of the requirements for the Degree of MASTER OF ENGINEERING (CHEMICAL ENGINEERING) in the Faculty of Engineering at Stellenbosch University Supervisor Prof. J.H. Knoetze Co-Supervisor/s Dr. C.E. Schwarz March 2015 Stellenbosch University https://scholar.sun.ac.za DECLARATION By submitting this thesis electronically, I declare that the entirety of the work contained therein is my own, original work, that I am the sole author thereof (save to the extent explicitly otherwise stated), that reproduction and publication thereof by Stellenbosch University will not infringe any third party rights and that I have not previously in its entirety or in part submitted it for obtaining any qualification. James Lombard February 2015 ………………………. ………………………. Signature Date Copyright © 2015 Stellenbosch University All rights reserved i Stellenbosch University https://scholar.sun.ac.za ABSTRACT Long-chain hydrocarbons are of value to numerous lucrative industries. Due to the low volatility and close melting and boiling points of these solutes, traditional fractionation methods lack the required selectivity for separation and cause thermal degradation of the product. This project investigates the feasibility of Supercritical Fluid Extraction (SFE) for processing these systems, with the primary objective of modelling the high-pressure vapour-liquid equilibrium (VLE) properties of hydrocarbon solutes with a light-weight solvent using a semi- empirical equation of state (EOS). Pure component vapour pressures and saturated liquid volumes are also investigated. A thorough investigation into the phase behaviour of the n-alkanes, 1-alcohols, carboxylic acids and esters in light weight supercritical solvents CO2, ethane and propane revealed that the solute structure and temperature largely influence the solute solubility and process feasibility. -

Summaries of FY 1991 Research in the Chemical Sciences
)Is - I~ )~i~TiI ~i*ii i~T -~~~~~~~~~~~~~~~~~~-l 9~~~~~~~~~~rsrr A S.~~~~~~~:Ir I... - - 0~Iii Available to DOE and DOE contractors from the Office of Scientific and Technical Information, P.O. Box 62, Oak Ridge, TN 37831; prices available from (615) 576-8401, FTS 626-8401. Available to the public from the National Technical Information Service, U.S. Department of Commerce, 5285 Port Royal Rd., Springfield, VA 22161. DOE/ER-01 44/9 (DE91011221) August 1991 Distribution Categories UC-400 and UC-401 Summaries of FY 1991 Research in the Chemical Sciences U.S. Department of Energy Office of Energy Research b^~~~~~~~~~~~~ ~~~~~Division of Chemical Sciences I This report was compiled for the Office of Energy Research from project summaries contained in the Research-In-Progress (RIP)database of the Office of Scientific and Technical Information, Oak Ridge, ; ^<-<~~~~~~~~~~~~~~ ~~Tennessee. The RIP database describes new and jr';(;-i~~~~~~~~ l ~~~~~~ongoingenergy and energy-related research projects carried out or sponsored by the Department of Energy. s .» \ Contents PREFACE vii University of Arizona 34 CHEMICAL SCIENCES DIVISION viii Boston University 34 PROGRAM SUMMARIES ix Brandeis University 34 LABORATORY ADMINISTRATION xi California Institute of Technology 35 University of California, Berkeley 35 NATIONAL LABORATORIES University of California, Irvine 35 Photochemical and Radiation Sciences University of California, Los Angeles 36 Ames Laboratory 1 University of California, Santa Barbara 36 Argonne National Laboratory 2 Clemson University -

Journal of Pharmaceutical and Pharmacological Sciences Samuel P, Et Al., J Pharma Pharma Sci: JPPS-138
Journal of Pharmaceutical and Pharmacological Sciences Samuel P, et al., J Pharma Pharma Sci: JPPS-138. Research Article DOI:10.29011/2574-7711/100038 Bioprospecting of Salicornia europaeaL. a Marine Halophyte and Evaluation of Its Biological Potential with Special Refer- ence to Anticancer Activity Samuel P1*, Vijaya J Kumar1, Deena R Dhayalan1, Amirtharaj K1, Sudarmani DNP2 1Department of Biotechnology, Ayya Nadar Janaki Ammal College(Autonomous), Sivakasi, TN, India 2Department of Zoology (PG), Ayya Nadar Janaki Ammal College (Autonomous), Sivakasi, TN, India *Corresponding author: Samuel P, Assistant Professor, Department of Biotechnology, Ayya Nadar Janaki Ammal College (Autono- mous), Sivakasi, TN, India. Tel: +919025559994; Email: [email protected] Citation: Samuel P, Kumar VJ, Dhayalan DR, Amirtharaj K, Sudarmani DNP (2017) Bioprospecting of Salicornia europaea L. a Marine Halophyte and Evaluation of Its Biological Potential with Special Reference to Anticancer Activity. J Pharma Pharma Sci: JPPS-138. DOI:10.29011/2574-7711/100038 Received Date: 02 August, 2017; Accepted Date: 25 August, 2017; Published Date: 02 September, 2017 Abstract The present study was initiated with an intention to bring outthe phytochemical profile of the marine halophyte Salicornia europaeaL., the chemical finger print of the halophyte was made by GC-MS.The phytochemicalsin the crude extract was evalu- ated for cytotoxic study against MCF7. The marine halophytes were collected, washedand chopped in to 5cm long and shade dried for 20-25 days in a dark room. The dried and grounded plant materials were subjected to Soxhlet extraction.Two solvents vizmethanol and ethyl acetate were used to prepare decoction of the plant.The extracts were screened using GC-MS and dried us- ing rotary vacuum evaporator. -

ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability Of
ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability of Coastal Plain Groundwater to Flood Water Intrusion using High Resolution Mass Spectrometry. (Under the direction of Dr. Elizabeth Guthrie Nichols). Communities in the North Carolina Coastal Plain (NCCP) depend on safe and reliable groundwater for private well use, agriculture, industry, and livelihoods. Although storm intensity and frequency are predicted to increase in coastal areas, the risk of surficial and confined aquifer contamination from extreme storms is not understood. In September 2018, Hurricane Florence caused extensive flooding across the NCCP for several weeks. The North Carolina Department of Environmental Quality (NCDEQ) Groundwater Management Branch had just completed sampling of some wells in their monitoring network when Hurricane Florence made landfall. NCDEQ returned to these wells, particularly those flooded by Hurricane Florence, for post-flood sampling. These groundwater samples were analyzed by NCDEQ for regulated semi-volatile organics with few to any detections of regulated organic contaminants. NCDEQ provided NC State the same sample extracts for analysis by high resolution mass spectrometry (HRMS). This research reports on the non-targeted and suspect-screening HRMS analyses of groundwater from nested monitoring wells. Some monitoring well sites experienced flooding during the study period, and some did not. The goal of this research was to advance our understanding of coastal aquifer susceptibility to flooding by producing the first comprehensive organic chemical profiles of coastal aquifers and by determining if aquifers have distinct organic chemical profiles that change after flooding events. This study used HRMS analyses to produce the first comprehensive organic chemical profiles of 11 aquifers in the coastal plain.