Alcohol-Responsive Genes Identified in Human Ipsc-Derived Neural Cultures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dehydrogenase Gene of Entamoeba Histolytica
Proc. Nad. Acad. Sci. USA Vol. 89, pp. 10188-10192, November 1992 Medical Sciences Cloning and expression of an NADP+-dependent alcohol dehydrogenase gene of Entamoeba histolytica (amebiasis/protozoan parasite/fermentation/inhibitors) AJIT KUMAR*, PEI-SHEN SHENtt, STEVEN DESCOTEAUX*, JAN POHL§, GORDON BAILEYt, AND JOHN SAMUELSON*1II *Department of Tropical Public Health, Harvard School of Public Health, Boston, MA 02115; tDepartment of Biochemistry, Morehouse School of Medicine, Atlanta, GA 30310; §Microchemical Facility, Emory University, Atlanta, GA 30322; and 'Department of Pathology, New England Deaconess Hospital, Boston, MA 02215 Communicated by Elkan Blout, June 29, 1992 (receivedfor review May 1, 1992) ABSTRACT Ethanol is the major metabolic product of NAD(P)+ so that the reducing equivalents are regenerated glucose fermentation by the protozoan parasite Entmoeba (4-7). Both NADP+-dependent and NAD+-dependent alco- histolytica under the anaerobic conditions found in the lumen hol dehydrogenase (ADH) activities have been identified in of the colon. Here an internal peptide sequence determined E. histolytica trophozoites (4-7). The NADP+-dependent from a major 39-kDa amoeba protein isolated by isoeLtric ADH (EhADHi; EC 1.1.1.2) was found to be composed of focusing followed by SDS/PAGE was used to clone the gene for four -30-kDa subunits, prefer secondary alcohols to primary the E. histolytica NADP+-dependent alcohol dehydrogenase alcohols, and be inhibited by the substrate analogue pyrazole (EhADH1; EC 1.1.1.2). The EhADHi clone had an open (7). The amoeba NAD+-dependent ADH (EC 1.1.1.1) is not reading frame that was 360 amino acids long and encoded a well-characterized (4). -

The Failure of Two Major Formaldehyde Catabolism Enzymes (ADH5 and ALDH2) Leads to Partial Synthetic Lethality in C57BL/ 6 Mice Jun Nakamura1,2*, Darcy W
Nakamura et al. Genes and Environment (2020) 42:21 https://doi.org/10.1186/s41021-020-00160-4 SHORT REPORT Open Access The failure of two major formaldehyde catabolism enzymes (ADH5 and ALDH2) leads to partial synthetic lethality in C57BL/ 6 mice Jun Nakamura1,2*, Darcy W. Holley3, Toshihiro Kawamoto4 and Scott J. Bultman3 Abstract Background: Exogenous formaldehyde is classified by the IARC as a Category 1 known human carcinogen. Meanwhile, a significant amount of endogenous formaldehyde is produced in the human body; as such, formaldehyde-derived DNA and protein adducts have been detected in animals and humans in the absence of major exogenous formaldehyde exposure. However, the toxicological effects of endogenous formaldehyde on individuals with normal DNA damage repair functions are not well understood. In this study, we attempted to generate C57BL/6 mice deficient in both Adh5 and Aldh2, which encode two major enzymes that metabolize endogenous formaldehyde, in order to understand the effects of endogenous formaldehyde on mice with normal DNA repair function. Results: Due to deficiencies in both ADH5 and ALDH2, few mice survived past post-natal day 21. In fact, the survival of pups within the first few days after birth was significantly decreased. Remarkably, two Aldh2−/−/Adh5−/− mice survived for 25 days after birth, and we measured their total body weight and organ weights. The body weight of Aldh2−/−/Adh5−/− mice decreased significantly by almost 37% compared to the Aldh2−/−/Adh5+/− and Aldh2−/−/Adh5+/+ mice of the same litter. In addition, the absolute weight of each organ was also significantly reduced. Conclusion: Mice deficient in both formaldehyde-metabolizing enzymes ADH5 and ALDH2 were found to develop partial synthetic lethality and mortality shortly after birth. -

Age-Dependent Protein Abundance of Cytosolic Alcohol and Aldehyde Dehydrogenases in Human Liver S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2017/08/21/dmd.117.076463.DC2 http://dmd.aspetjournals.org/content/suppl/2017/06/12/dmd.117.076463.DC1 1521-009X/45/9/1044–1048$25.00 https://doi.org/10.1124/dmd.117.076463 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 45:1044–1048, September 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics Age-dependent Protein Abundance of Cytosolic Alcohol and Aldehyde Dehydrogenases in Human Liver s Deepak Kumar Bhatt, Andrea Gaedigk, Robin E. Pearce, J. Steven Leeder, and Bhagwat Prasad Department of Pharmaceutics, University of Washington, Seattle, Washington (D.K.B., B.P.); Department of Clinical Pharmacology, Toxicology & Therapeutic Innovation, Children’s Mercy-Kansas City, Missouri and School of Medicine, University of Missouri-Kansas City, Kansas City, Missouri (A.G., R.E.P., J.S.L.) Received April 21, 2017; accepted June 5, 2017 ABSTRACT Hepatic cytosolic alcohol and aldehyde dehydrogenases (ADHs and the adult levels, respectively. For all proteins, the abundance steeply ALDHs) catalyze the biotransformation of xenobiotics (e.g., cyclo- increased during the first year of life, which mostly reached adult Downloaded from phosphamide and ethanol) and vitamin A. Because age-dependent levels during early childhood (age between 1 and 6 years). Only for hepatic abundance of these proteins is unknown, we quantified ADH1A protein abundance in adults (age > 18 year) was ∼40% lower protein expression of ADHs and ALDH1A1 in a large cohort of pediatric relative to the early childhood group. Abundances of ADHs and and adult human livers by liquid chromatography coupled with tandem ALDH1A1 were not associated with sex in samples with age > 1 year mass spectrometry proteomics. -

Confirmation of Pathogenic Mechanisms by SARS-Cov-2–Host
Messina et al. Cell Death and Disease (2021) 12:788 https://doi.org/10.1038/s41419-021-03881-8 Cell Death & Disease ARTICLE Open Access Looking for pathways related to COVID-19: confirmation of pathogenic mechanisms by SARS-CoV-2–host interactome Francesco Messina 1, Emanuela Giombini1, Chiara Montaldo1, Ashish Arunkumar Sharma2, Antonio Zoccoli3, Rafick-Pierre Sekaly2, Franco Locatelli4, Alimuddin Zumla5, Markus Maeurer6,7, Maria R. Capobianchi1, Francesco Nicola Lauria1 and Giuseppe Ippolito 1 Abstract In the last months, many studies have clearly described several mechanisms of SARS-CoV-2 infection at cell and tissue level, but the mechanisms of interaction between host and SARS-CoV-2, determining the grade of COVID-19 severity, are still unknown. We provide a network analysis on protein–protein interactions (PPI) between viral and host proteins to better identify host biological responses, induced by both whole proteome of SARS-CoV-2 and specific viral proteins. A host-virus interactome was inferred, applying an explorative algorithm (Random Walk with Restart, RWR) triggered by 28 proteins of SARS-CoV-2. The analysis of PPI allowed to estimate the distribution of SARS-CoV-2 proteins in the host cell. Interactome built around one single viral protein allowed to define a different response, underlining as ORF8 and ORF3a modulated cardiovascular diseases and pro-inflammatory pathways, respectively. Finally, the network-based approach highlighted a possible direct action of ORF3a and NS7b to enhancing Bradykinin Storm. This network-based representation of SARS-CoV-2 infection could be a framework for pathogenic evaluation of specific 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; clinical outcomes. -

How Is Alcohol Metabolized by the Body?
Overview: How Is Alcohol Metabolized by the Body? Samir Zakhari, Ph.D. Alcohol is eliminated from the body by various metabolic mechanisms. The primary enzymes involved are aldehyde dehydrogenase (ALDH), alcohol dehydrogenase (ADH), cytochrome P450 (CYP2E1), and catalase. Variations in the genes for these enzymes have been found to influence alcohol consumption, alcohol-related tissue damage, and alcohol dependence. The consequences of alcohol metabolism include oxygen deficits (i.e., hypoxia) in the liver; interaction between alcohol metabolism byproducts and other cell components, resulting in the formation of harmful compounds (i.e., adducts); formation of highly reactive oxygen-containing molecules (i.e., reactive oxygen species [ROS]) that can damage other cell components; changes in the ratio of NADH to NAD+ (i.e., the cell’s redox state); tissue damage; fetal damage; impairment of other metabolic processes; cancer; and medication interactions. Several issues related to alcohol metabolism require further research. KEY WORDS: Ethanol-to acetaldehyde metabolism; alcohol dehydrogenase (ADH); aldehyde dehydrogenase (ALDH); acetaldehyde; acetate; cytochrome P450 2E1 (CYP2E1); catalase; reactive oxygen species (ROS); blood alcohol concentration (BAC); liver; stomach; brain; fetal alcohol effects; genetics and heredity; ethnic group; hypoxia The alcohol elimination rate varies state of liver cells. Chronic alcohol con- he effects of alcohol (i.e., ethanol) widely (i.e., three-fold) among individ- sumption and alcohol metabolism are on various tissues depend on its uals and is influenced by factors such as strongly linked to several pathological concentration in the blood T chronic alcohol consumption, diet, age, consequences and tissue damage. (blood alcohol concentration [BAC]) smoking, and time of day (Bennion and Understanding the balance of alcohol’s over time. -

Computational Exploration of the Medium-Chain Dehydrogenase / Reductase Superfamily
From the Department of Medical Biochemistry and Biophysics Karolinska Institutet, Stockholm, Sweden COMPUTATIONAL EXPLORATION OF THE MEDIUM-CHAIN DEHYDROGENASE / REDUCTASE SUPERFAMILY Linus J. Östberg Stockholm 2017 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by E-Print AB 2017 ©Linus J. Östberg, 2017 ISBN 978-91-7676-650-7 COMPUTATIONAL EXPLORATION OF THE MEDIUM-CHAIN DEHYDROGENASE / REDUCTASE SUPERFAMILY THESIS FOR DOCTORAL DEGREE (Ph.D.) By Linus J. Östberg Principal Supervisor: Opponent: Prof. Jan-Olov Höög Prof. Jaume Farrés Karolinska Institutet Autonomous University of Barcelona Department of Medical Biochemistry and Department of Biochemistry and Biophysics Molecular Biology Co-supervisor: Examination Board: Prof. Bengt Persson Prof. Erik Lindahl Uppsala University Stockholm University Department of Cell and Molecular Biology Department of Biochemistry and Biophysics Assoc. Prof. Tanja Slotte Stockholm University Department of Ecology, Environment and Plant Sciences Prof. Elias Arnér Karolinska Institutet Department of Medical Biochemistry and Biophysics ABSTRACT The medium-chain dehydrogenase/reductase (MDR) superfamily is a protein family with more than 170,000 members across all phylogenetic branches. In humans there are 18 repre- sentatives. The entire MDR superfamily contains many protein families such as alcohol dehy- drogenase, which in mammals is in turn divided into six classes, class I–VI (ADH1–6). Most MDRs have enzymatical functions, catalysing the conversion of alcohols to aldehyde/ketones and vice versa, but the function of some members is still unknown. In the first project, a methodology for identifying and automating the classification of mammalian ADHs was developed using BLAST for identification and class-specific hidden Markov models were generated for identification. -

ARTICLE Evidence of Positive Selection on a Class I ADH Locus
ARTICLE Evidence of Positive Selection on a Class I ADH Locus Yi Han,* Sheng Gu,* Hiroki Oota,† Michael V. Osier,‡ Andrew J. Pakstis, William C. Speed, Judith R. Kidd, and Kenneth K. Kidd The alcohol dehydrogenase (ADH) family of enzymes catalyzes the reversible oxidation of alcohol to acetaldehyde. Seven ADH genes exist in a segment of ∼370 kb on 4q21. Products of the three class I ADH genes that share 95% sequence identity are believed to play the major role in the first step of ethanol metabolism. Because the common belief that selection has operated at the ADH1B*47His allele in East Asian populations lacks direct biological or statistical evidence, we used genomic data to test the hypothesis. Data consisted of 54 single-nucleotide polymorphisms (SNPs) across the ADH clusters in a global sampling of 42 populations. Both the Fst statistic and the long-range haplotype (LRH) test provided positive evidence of selection in several East Asian populations. The ADH1B Arg47His functional polymorphism has the highest Fst of the 54 SNPs in the ADH cluster, and it is significantly above the mean Fst of 382 presumably neutral sites tested on the same 42 population samples. The LRH test that uses cores including that site and extending on both sides also gives significant evidence of positive selection in some East Asian populations for a specific haplotype carrying the ADH1B*47His allele. Interestingly, this haplotype is present at a high frequency in only some East Asian populations, whereas the specific allele also exists in other East Asian populations and in the Near East and Europe but does not show evidence of selection with use of the LRH test. -

Anti-ADH5 Monoclonal Antibody, Clone FQS23996(C) (DCABH-6425) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-ADH5 monoclonal antibody, clone FQS23996(C) (DCABH-6425) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Rabbit monoclonal to ADH5 Antigen Description Class-III ADH is remarkably ineffective in oxidizing ethanol, but it readily catalyzes the oxidation of long-chain primary alcohols and the oxidation of S-(hydroxymethyl) glutathione. Immunogen Synthetic peptide (the amino acid sequence is considered to be commercially sensitive) within Human ADH5 aa 1-100. The exact sequence is proprietary.Database link: P11766 Isotype IgG Source/Host Rabbit Species Reactivity Mouse, Rat, Human Clone FQS23996(C) Purity Tissue culture supernatant Conjugate Unconjugated Applications WB, IHC-P, IP, Flow Cyt Positive Control Human fetal liver, HepG2, Human fetal kidney and SH-SY5Y lysates. Human brain and kidney tissue. Permeabilized SH-SY5Y cells. Immunoprecipitation pellet from Human fetal liver lysate. Format Liquid Size 100 μl Buffer pH: 7.2; Preservative: 0.01% Sodium azide; Constituents: 49% PBS, 50% Glycerol, 0.05% BSA Preservative 0.01% Sodium Azide Storage Store at +4°C short term (1-2 weeks). Upon delivery aliquot. Store at -20°C long term. Avoid freeze / thaw cycle. Ship Shipped at 4°C. GENE INFORMATION 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Gene Name ADH5 alcohol dehydrogenase 5 (class III), chi polypeptide [ Homo sapiens ] Official Symbol ADH5 Synonyms -

A Curated Biochemical Pathways Database for the Laboratory Mouse Alexei V Evsikov, Mary E Dolan, Michael P Genrich, Emily Patek and Carol J Bult
Open Access Software2009EvsikovetVolume al. 10, Issue 8, Article R84 MouseCyc: a curated biochemical pathways database for the laboratory mouse Alexei V Evsikov, Mary E Dolan, Michael P Genrich, Emily Patek and Carol J Bult Address: The Jackson Laboratory, Main Street, Bar Harbor, ME 04609, USA. Correspondence: Carol J Bult. Email: [email protected] Published: 14 August 2009 Received: 22 May 2009 Revised: 17 July 2009 Genome Biology 2009, 10:R84 (doi:10.1186/gb-2009-10-8-r84) Accepted: 14 August 2009 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2009/10/8/R84 © 2009 Evsikov et al,; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. MouseCyc<p>MouseCyc database is a database of curated metabolic pathways for the laboratory mouse.</p> Abstract Linking biochemical genetic data to the reference genome for the laboratory mouse is important for comparative physiology and for developing mouse models of human biology and disease. We describe here a new database of curated metabolic pathways for the laboratory mouse called MouseCyc http://mousecyc.jax.org. MouseCyc has been integrated with genetic and genomic data for the laboratory mouse available from the Mouse Genome Informatics database and with pathway data from other organisms, including human. Rationale Biochemical interactions and transformations among organic The availability of the nearly complete genome sequence for molecules are arguably the foundation and core distinguish- the laboratory mouse provides a powerful platform for pre- ing feature of all organic life. -

32-2124: ADH5 Recombinant Protein Description Product Info
9853 Pacific Heights Blvd. Suite D. San Diego, CA 92121, USA Tel: 858-263-4982 Email: [email protected] 32-2124: ADH5 Recombinant Protein Alcohol dehydrogenase 5 (class III) chi polypeptide,Alcohol dehydrogenase class chi chain,Glutathione- Alternative dependent formaldehyde dehydrogenase,S-(hydroxymethyl) glutathione Name : dehydrogenase,FDH,ADHX,ADH-3,FALDH,GSH-FDH,GSNOR,EC 1.1.1.1,EC 1.1.1.2 Description Source : E.coli. ADH5 Recombinant produced in E. coli is a single polypeptide chain containing 398 amino acids (1-374) and having a molecular mass of 42.3kDa.ADH5 is fused to a 24 amino acid His-tag at N-terminus & purified by proprietary chromatographic techniques. ADH5 belongs to the alcohol dehydrogenase family which metabolizes a large selection of substrates, such as retinol, ethanol, other aliphatic alcohols, hydroxysteroids, and lipid peroxidation products. ADH5 has practically no activity for ethanol oxidation, but displays high activity for oxidation of long-chain primary alcohols and for oxidation of S-hydroxymethyl-glutathione, a spontaneous adduct between glutathione and formaldehyde. ADH5 enzyme is a key factor of cellular metabolism for the removal of formaldehyde, a powerful aggravating and alerting mediator which causes pharyngitis, lacrymation, rhinitis and contact dermatitis. Product Info Amount : 20 µg Purification : Greater than 90% as determined by SDS-PAGE. The ADH5 solution (1mg/ml) contains 20mM Tris-HCl buffer (pH 8.0), 100mM NaCl, 1mM DTT Content : and 20% glycerol. Store at 4°C if entire vial will be used within 2-4 weeks. Store, frozen at -20°C for longer periods of Storage condition : time. For long term storage it is recommended to add a carrier protein (0.1% HSA or BSA).Avoid multiple freeze-thaw cycles. -
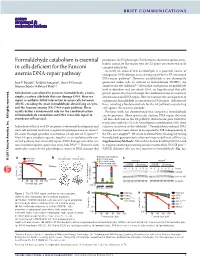
Formaldehyde Catabolism Is Essential in Cells Deficient for the Fanconi
BRIEF COMMUNICATIONS Formaldehyde catabolism is essential precipitates the FA phenotype. Furthermore, chemotherapeutic cross- linkers cannot be the reason why the FA genes are conserved in all in cells deficient for the Fanconi complex eukaryotes. Recently we showed that acetaldehyde is a potential source of anemia DNA-repair pathway endogenous DNA damage necessitating repair by the FA-associated DNA-repair pathway9. However, acetaldehyde is not abundantly Ivan V Rosado1, Frédéric Langevin1, Gerry P Crossan1, generated within cells, in contrast to formaldehyde (HCHO), the Minoru Takata2 & Ketan J Patel1,3 simplest reactive aldehyde10. Given that endogenous formaldehyde both is abundant and can attack DNA, we hypothesized that cells Metabolism is predicted to generate formaldehyde, a toxic, protect against this threat through the combined action of enzymatic simple, reactive aldehyde that can damage DNA. Here we detoxification and DNA repair. Here we examine the consequences of report a synthetic lethal interaction in avian cells between endogenous formaldehyde accumulation in DNA repair–deficient cell ADH5, encoding the main formaldehyde-detoxifying enzyme, lines, revealing a fundamental role for the FA pathway in protecting and the Fanconi anemia (FA) DNA-repair pathway. These cells against this reactive aldehyde. results define a fundamental role for the combined action Previous work has demonstrated that exogenous formaldehyde of formaldehyde catabolism and DNA cross-link repair in can be genotoxic. More specifically, chicken DNA repair–deficient All rights reserved. vertebrate cell survival. cell lines deficient in the FA pathway downstream gene FANCD2, translesion synthesis (TLS) or homologous recombination (HR) show Inc Individuals afflicted with FA are prone to abnormal development and selective sensitivity to this aldehyde11. -

Genetic Variants in Drosophila Disguised by Cofactor and Subunit Binding (Electrophoresis/Polymorphism/Alcohol Dehydrogenase/NAD) GEORGE B
Proc. Nati. Acad. Sci. USA Vol. 75, No. 1, pp. 395-399, January 1978 Genetics Structural flexibility of isozyme variants: Genetic variants in Drosophila disguised by cofactor and subunit binding (electrophoresis/polymorphism/alcohol dehydrogenase/NAD) GEORGE B. JOHNSON Department of Biology, Washington University, St. Louis, Missouri 63130 Communicated by Peter H. Raven, September 16, 1977 ABSTRACT Wild populations of Drosophila mojavensis cause any factor that tends to stabilize tertiary structure will exhibit considerable conformational variation in the NAD+-free act to obscure the variation from detection by electrophoresis. form of alcohol dehydrogenase (alcohol:NAD+ oxidoreductase; Thus, enzymes that tightly bind cofactors or that aggregate into EC 1.1.1.1). The variation appears genetic, as it does not occur within an inbred strain. The NAD+-bound form of alcohol de- multimers may exhibit fewer conformational classes than might hydrogenase, present in the same individuals, does not exhibit have been detected in the absence of the stabilizing influences the variation, suggesting that the binding of NAD+ acts to sta- of cofactor or subunit binding. bilize conformation. Such cofactor binding to enzymes may thus This paper reports studies on the stabilizing influences of conceal considerable variation. A similar effect is suggested for cofactor binding and of subunit aggregation on enzyme shape binding of esterase subunits. in Drosophila. The results suggest that variation in protein In the decade since electrophoretic variation at enzyme loci was shape may indeed be masked by such stabilizing influences. first reported (1), extensive investigation of insect (2), plant (3), MATERIALS AND METHODS small vertebrate (4), and human (5) populations has confirmed the generality of widespread polymorphism.