Pharmacokinetics JEFFERY A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1: Clinical Pharmacokinetics 1
1: CLINICAL PHARMACOKINETICS 1 General overview: clinical pharmacokinetics, 2 Pharmacokinetics, 4 Drug clearance (CL), 6 Volume of distribution (Vd), 8 The half-life (t½), 10 Oral availability (F), 12 Protein binding (PB), 14 pH and pharmacokinetics, 16 1 Clinical pharmacokinetics General overview General overview: clinical pharmacokinetics 1 The ultimate aim of drug therapy is to achieve effi cacy without toxicity. This involves achieving a plasma concentration (Cp) within the ‘therapeutic window’, i.e. above the min- imal effective concentration (MEC), but below the minimal toxic concentration (MTC). Clinical pharmacokinetics is about all the factors that determine variability in the Cp and its time-course. The various factors are dealt with in subsequent chapters. Ideal therapeutics: effi cacy without toxicity Minimum Toxic Concentration (MTC) Ideal dosing Minimum Effective Concentration (MEC) Drug concentration Time The graph shows a continuous IV infusion at steady state, where the dose-rate is exactly appropriate for the patient’s clearance (CL). Inappropriate dosing Dosing too high in relation to the patient’s CL – toxicity likely Minimum Toxic Concentration (MTC) Minimum Effective Concentration (MEC) Dosing too low in relation to the Drug concentration patient’s CL – drug may be ineffective Time Some reasons for variation in CL Low CL High CL Normal variation Normal variation Renal impairment Increased renal blood fl ow Genetic poor metabolism Genetic hypermetabolism Liver impairment Enzyme induction Enzyme inhibition Old age/neonate 2 General overview Clinical Pharmacokinetics Pharmacokinetic factors determining ideal therapeutics If immediate effect is needed, a loading dose (LD) must be given to achieve a desired 1 concentration. The LD is determined by the volume of distribution (Vd). -

Narrow Therapeutic Index
Therapeutic Drug Monitoring Some drugs have a narrow therapeutic index, which means that there is only a small difference between the minimum effective concentrations and the minimum toxic concentrations in the blood. With such drugs, small increases in dose or in blood/serum concentrations could lead to toxic effects. Therapeutic drug monitoring may help to optimise treatment in cases where there is a firm relationship between the toxic/therapeutic effects and drug concentrations in whole blood/serum. A therapeutic interval has been defined for the drugs in the following tables. This is the minimum effective and maximum safe concentration for compliant patients, on stabilised regimens. Levels within these limits should prove satisfactory in most cases. Whole blood or serum drug concentrations are useful for determining patient compliance or for assessing whether or not: 1. adequate concentrations are being achieved or, 2. potentially toxic concentrations are being reached. Depending on clinical conditions, dosage adjustments may be needed when levels are outside the therapeutic interval. Therapeutic drug monitoring can also be useful when changes are made to other medications that could affect serum or whole blood concentrations and lead to a reduction in effectiveness or increased toxicity. Although plasma drug concentrations and the therapeutic interval are useful in evaluating drug therapy, they should not be the only criteria on which treatment is based. Always remember to treat the patient, not the level. Drug concentrations in serum or whole blood are only meaningful if the correct procedures are followed regarding the timing of specimens and selection of sample tube. It is vitally important to note the exact time the sample is taken and when each dose of the drug is given. -

Narrow Therapeutic Index Drugs: a Clinical Pharmacological Consideration to Flecainide Juan Tamargo, Jean-Yves Le Heuzey, Phillipe Mabo
Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide Juan Tamargo, Jean-Yves Le Heuzey, Phillipe Mabo To cite this version: Juan Tamargo, Jean-Yves Le Heuzey, Phillipe Mabo. Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide. European Journal of Clinical Pharmacology, Springer Verlag, 2015, 71 (5), pp.549-567. 10.1007/s00228-015-1832-0. hal-01143095 HAL Id: hal-01143095 https://hal-univ-rennes1.archives-ouvertes.fr/hal-01143095 Submitted on 16 Apr 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Eur J Clin Pharmacol (2015) 71:549–567 DOI 10.1007/s00228-015-1832-0 REVIEW ARTICLE Narrow therapeutic index drugs: a clinical pharmacological consideration to flecainide Juan Tamargo & Jean-Yves Le Heuzey & Phillipe Mabo Received: 5 December 2014 /Accepted: 4 March 2015 /Published online: 15 April 2015 # The Author(s) 2015. This article is published with open access at Springerlink.com Abstract specify flecainide as an NTID. The literature review demon- Purpose The therapeutic index (TI) is the range of doses at strated that flecainide displays NTID characteristics including which a medication is effective without unacceptable adverse a steep drug dose–response relationship for safety and effica- events. -

Pharmacogenetic Testing: a Tool for Personalized Drug Therapy Optimization
pharmaceutics Review Pharmacogenetic Testing: A Tool for Personalized Drug Therapy Optimization Kristina A. Malsagova 1,* , Tatyana V. Butkova 1 , Arthur T. Kopylov 1 , Alexander A. Izotov 1, Natalia V. Potoldykova 2, Dmitry V. Enikeev 2, Vagarshak Grigoryan 2, Alexander Tarasov 3, Alexander A. Stepanov 1 and Anna L. Kaysheva 1 1 Biobanking Group, Branch of Institute of Biomedical Chemistry “Scientific and Education Center”, 109028 Moscow, Russia; [email protected] (T.V.B.); [email protected] (A.T.K.); [email protected] (A.A.I.); [email protected] (A.A.S.); [email protected] (A.L.K.) 2 Institute of Urology and Reproductive Health, Sechenov University, 119992 Moscow, Russia; [email protected] (N.V.P.); [email protected] (D.V.E.); [email protected] (V.G.) 3 Institute of Linguistics and Intercultural Communication, Sechenov University, 119992 Moscow, Russia; [email protected] * Correspondence: [email protected]; Tel.: +7-499-764-9878 Received: 2 November 2020; Accepted: 17 December 2020; Published: 19 December 2020 Abstract: Pharmacogenomics is a study of how the genome background is associated with drug resistance and how therapy strategy can be modified for a certain person to achieve benefit. The pharmacogenomics (PGx) testing becomes of great opportunity for physicians to make the proper decision regarding each non-trivial patient that does not respond to therapy. Although pharmacogenomics has become of growing interest to the healthcare market during the past five to ten years the exact mechanisms linking the genetic polymorphisms and observable responses to drug therapy are not always clear. Therefore, the success of PGx testing depends on the physician’s ability to understand the obtained results in a standardized way for each particular patient. -

Narrow Therapeutic Index Drugs
Quality and Bioequivalence Standards for Narrow Therapeutic Index Drugs Lawrence X. Yu, Ph.D. Deputy Director for Science and Chemistry Office of Generic Drugs Center for Drug Evaluation and Research Food and Drug Administration GPhA 2011 Fall Technical Workshop 1 Bioequivalence • The absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administrated at the same molar dose under similar conditions in an appropriately designed study…” (21 CFR §320.1) 2 Plasma Concentration Profile Cmax 10000 AUC ln Concentration 1000 Concentration Time Tmax - time of maximum concentration 100 Time 3 0 5 10 15 20 25 Possible Outcome of BE Studies Demonstrate BE Fail to Demonstrate BIE Fail to Demonstrate BE Demonstrate BIE Demonstrate BIE 80% T/R (%) 125% 4 5 FDA 12 Year BE Data Distribution of AUCt Ratios Average difference = 3.56% 10 N = 2069 8 6 Percent of Total(%) of Percent 4 2 0 0.84 0.86 0.88 0.90 0.92 0.94 0.96 0.98 1.00 1.02 1.04 1.06 1.08 1.10 1.12 1.14 1.16 1.18 1.20 6 AUC Point Estimate (T/R) Effect of Variability on BE Studies High variability 80% T/R (%) 125% 7 Development of BE Standard for Highly Variable Drugs 4/2004 First presentation to the FDA Advisory Committee 10/2006 Second presentation to the FDA Advisory Committee 3/2007 Received the first ANDA which used the new FDA BE approach 5/2007 Critical Path Opportunities for Generic Drugs BE of HVD 1/2008 FDA OGD’s first publication on BE of HVD (Pharm. -

Warfarin Sodium
Contains Nonbinding Recommendations Draft Guidance on Warfarin Sodium This draft guidance, once finalized, will represent the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the Office of Generic Drugs. Active ingredient: Warfarin Sodium Form/Route: Tablet/Oral Recommended studies: 2 studies 1. Type of study: Fasting Design: 4-way, fully replicated crossover design in-vivo Strength: 10 mg Subjects: Healthy males and nonpregnant females, general population. Additional Comments: Warfarin has a long terminal elimination half-life. Please ensure adequate washout periods between treatments in the crossover studies. For long half-life drug products, an AUC truncated to 72 hours may be used in place of AUC0-t or AUC0-∞, as described in the Guidance for Industry: “Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations”. Applicants may consider using the reference-scaled average bioequivalence approach for warfarin described below. 2. Type of study: Fed Design: 4-way, fully replicated crossover design in-vivo Strength: 10 mg Subjects: Healthy males and nonpregnant females, general population. Additional Comments: See additional comments above. See Amantadine Hydrochloride Oral Tablet guidance -

Anew Drug Design Strategy in the Liht of Molecular Hybridization Concept
www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 “Drug Design strategy and chemical process maximization in the light of Molecular Hybridization Concept.” Subhasis Basu, Ph D Registration No: VB 1198 of 2018-2019. Department Of Chemistry, Visva-Bharati University A Draft Thesis is submitted for the partial fulfilment of PhD in Chemistry Thesis/Degree proceeding. DECLARATION I Certify that a. The Work contained in this thesis is original and has been done by me under the guidance of my supervisor. b. The work has not been submitted to any other Institute for any degree or diploma. c. I have followed the guidelines provided by the Institute in preparing the thesis. d. I have conformed to the norms and guidelines given in the Ethical Code of Conduct of the Institute. e. Whenever I have used materials (data, theoretical analysis, figures and text) from other sources, I have given due credit to them by citing them in the text of the thesis and giving their details in the references. Further, I have taken permission from the copyright owners of the sources, whenever necessary. IJCRT2012039 International Journal of Creative Research Thoughts (IJCRT) www.ijcrt.org 284 www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 f. Whenever I have quoted written materials from other sources I have put them under quotation marks and given due credit to the sources by citing them and giving required details in the references. (Subhasis Basu) ACKNOWLEDGEMENT This preface is to extend an appreciation to all those individuals who with their generous co- operation guided us in every aspect to make this design and drawing successful. -

Guidance for Industry: Bioavailability and Bioequivalence Studies For
Guidance for Industry Bioavailability and Bioequivalence Studies for Orally Administered Drug Products — General Considerations DRAFT GUIDANCE This guidance document is being distributed for comment purposes only. Comments and suggestions regarding this draft document should be submitted within 30 days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit comments to Dockets Management Branch (HFA-305), Food and Drug Administration, 5630 Fishers Lane, rm. 1061, Rockville, MD 20852. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register. For questions regarding this draft document contact (CDER) Aida Sanchez 301-827-5847. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) July 2002 BP J:\!GUIDANC\4964dft.doc 07/10/02 Guidance for Industry Bioavailability and Bioequivalence Studies for Orally Administered Drug Products — General Considerations Additional copies are available from: Office of Training and Communication Division of Drug Information, HFD-240 Center for Drug Evaluation and Research Food and Drug Administration 5600 Fishers Lane Rockville, MD 20857 (Tel) 301-827-4573 http://www.fda.gov/cder/guidance/index.htm U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) July 2002 BP J:\!GUIDANC\4964dft.doc 07/10/02 TABLE OF CONTENTS I. INTRODUCTION................................................................................................................ -
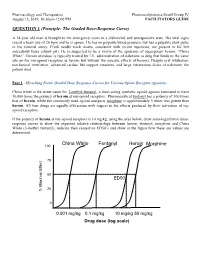
QUESTION 1 (Principle: the Graded Dose-Response Curve)
Pharmacology and Therapeutics Pharmacodynamics Small Group IV August 15, 2019, 10:30am-12:00 PM FACILITATORS GUIDE QUESTION 1 (Principle: The Graded Dose-Response Curve) A 34-year old man is brought to the emergency room in a disheveled and unresponsive state. His vital signs reveal a heart rate of 26 bpm and he is apneic. He has no palpable blood pressure, but has a palpable slow pulse in his femoral artery. Fresh needle track marks, consistent with recent injections, are present in his left antecubital fossa (elbow pit). He is suspected to be a victim of the epidemic of superpotent heroin, “China White”. Heroin overdose is typically treated by I.V. administration of naloxone (a drug that binds to the same site on the mu-opioid receptors as heroin, but without the narcotic effects of heroin). Despite oral intubation, mechanical ventilation, advanced cardiac life support measures, and large intravenous doses of naloxone, the patient died. Part 1 (Teaching Point: Graded Dose Response Curves for Various Opiate Receptor Agonists) China white is the street name for 3-methyl-fentanyl, a short-acting synthetic opioid agonist estimated to have 10,000 times the potency of heroin at mu-opioid receptors. Pharmaceutical fentanyl has a potency of 100 times that of heroin, while the commonly used opioid analgesic morphine is approximately 5 times less potent than heroin. All four drugs are equally efficacious with respect to the effects produced by their activation of m opioid receptors. If the potency of heroin at mu-opioid receptors is 10 mg/kg, using the axes below, draw semi-logarithmic dose- response curves to show the expected relative relationships between heroin, fentanyl, morphine and China White (3-methyl fentanyl), indicate their respective ED50’s and show in the figure how these are values are determined. -

Drugs with Narrow Therapeutic Index As Indicators in the Risk Management of Hospitalised Patients
Blix HS, Viktil KK, Moger TA, Reikvam A. Drugs with narrow therapeutic index as indicators in the risk management of hospitalised patients. Pharmacy Practice (Internet) 2010 Jan-Mar;8(1):50-55. Original Research Drugs with narrow therapeutic index as indicators in the risk management of hospitalised patients Hege S. BLIX, Kirsten K. VIKTIL, Tron A. MOGER, Aasmund REIKVAM. Received (first version): 11-Sep-2009 Accepted: 18-Jan-2010 ABSTRACT* is a well-suited tool for characterising the risk Drugs with narrow therapeutic index (NTI-drugs) are attributed to various drugs. drugs with small differences between therapeutic and toxic doses. The pattern of drug-related Keywords: Clinical Pharmacy Information Systems. problems (DRPs) associated with these drugs has Drug Toxicity. Inpatients. Norway. not been explored. Objective: To investigate how, and to what extent drugs, with a narrow therapeutic index (NTI-drugs), MEDICAMENTOS DE MARGEN as compared with other drugs, relate to different TERAPÉUTICO ESTRECHO COMO types of drug-related problems (DRPs) in INDICADORES DE GESTIÓN DE RIESGO hospitalised patients. Methods: Patients from internal medicine and EN PACIENTES HOSPITALIZADOS rheumatology departments in five Norwegian hospitals were prospectively included in 2002. RESUMEN Clinical pharmacists recorded demographic data, Los medicamentos con estrecho margen terapéutico drugs used, medical history and laboratory data. (NTI) son medicamentos con pequeñas diferencias Patients who used NTI-drugs (aminoglycosides, entre las dosis terapéuticas y tóxicas. No se han ciclosporin, carbamazepine, digoxin, digitoxin, explorado los problemas relacionados con flecainide, lithium, phenytoin, phenobarbital, medicamentos (DRPs) de estos medicamentos. rifampicin, theophylline, warfarin) were compared Objetivo: Investigar cómo y cuanto se relacionan with patients not using NTI-drugs. -

Development Team
Drug Concentration and Therapeutic Response Development Team Principal Investigator Prof. Farhan J Ahmad Jamia Hamdard, New Delhi Paper Coordinator Dr. Javed Ali Jamia Hamdard, New Delhi Dr. Shadab Md. School of Pharmacy, Content Writer International Medical University (IMU), Kuala Lumpur, Malaysia Dr. Sonal Gupta, KL Mehta Dayanand college, Content Reviewer Faridabad Pharmaceutical Biopharmaceutics and Pharmacokinetics sciences Drug Concentration and Therapeutic Response 0 Drug Concentration and Therapeutic Response Drug Concentration and Therapeutic Response Content 1. Introduction 2. Dose/Concentration response relationship 3. Types of dose and response relationship 3.1. Potency 3.2. Efficacy 3.3. Selectivity 3.4. Affinity 4. Therapeutic Window 4.1. Therapeutic Index 5. The relationship between drug concentration and pharmacological effects in the whole animal/human 6. Pharmacodynamic model 7. Onset and Duration of Action Pharmaceutical Biopharmaceutics and Pharmacokinetics sciences Drug Concentration and Therapeutic Response 1 Drug Concentration and Therapeutic Response I. Introduction To produce therapeutic/beneficial effect or minor, major, serious and severe toxic effects, drugs interact with receptors, ion channels, membrane carriers or enzymes in the body; this is called pharmacodynamics action of drug. The drug-receptor interactions usually occur in the tissue which is in equilibrium with the unbound drug (not bound to the plasma protein) present in the plasma. Drugs bind and interact in a structurally specific manner with these protein receptors. Activation of receptors in response to drug binding leads to activation of a second messenger system, resulting in a physiological or biochemical response such as changes in intracellular calcium concentrations leading to muscle contraction or relaxation. There are four main types of receptor families i. -

Principles of Pharmacology
© Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC manning/Shutterstock © Sofi NOT FOR SALE OR DISTRIBUTION NOT FOR2 SALE OR DISTRIBUTION Principles of Pharmacology © Jones & Bartlett Learning, LLC Laura Williford Owens,© Robin Jones Webb & Corbett, Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTIONTekoa L. King NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE✜ Chapter OR DISTRIBUTION Glossary NOT FOR SALE OR DISTRIBUTION exchange of drugs between the systemic circulation Absorption Movement of drug particles from the gastro- and the circulation in the central nervous system. intestinal tract to the systemic circulation by passive Chronobiology (chronopharmacology) Use of knowledge of cir- absorption, active transport, or pinocytosis. cadian rhythms to time administration of drugs for © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC Affinity Degree of attraction between a drug and a recep- maximum benefi t and minimal harm. tor. Th e greater NOTthe attraction, FOR SALE the greater OR DISTRIBUTIONthe extent Clearance Measure of the body’sNOT ability FOR to SALE eliminate OR a drug.DISTRIBUTION of binding. Competitive antagonist Drug or ligand that reversibly binds Agonist Drug that activates a receptor when bound to that to receptors at the same receptor site that agonists use receptor. (active site) without activating the receptor to initiate Agonist–antagonist© Jones & BartlettDrug that Learning,has agonist properties LLC for one a reaction.© Jones & Bartlett Learning, LLC NOTopioid FOR receptor SALE and antagonist OR DISTRIBUTION properties for a diff erent CytochromeNOT P-450 FOR (CYP450) SALE Generic OR name DISTRIBUTION for the family of type of opioid receptor.