Molecular Techniques for the Study of Helicobacter Sp
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Food Or Beverage Product, Or Probiotic Composition, Comprising Lactobacillus Johnsonii 456
(19) TZZ¥¥¥ _T (11) EP 3 536 328 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 11.09.2019 Bulletin 2019/37 A61K 35/74 (2015.01) A61K 35/66 (2015.01) A61P 35/00 (2006.01) (21) Application number: 19165418.5 (22) Date of filing: 19.02.2014 (84) Designated Contracting States: • SCHIESTL, Robert, H. AL AT BE BG CH CY CZ DE DK EE ES FI FR GB Encino, CA California 91436 (US) GR HR HU IE IS IT LI LT LU LV MC MK MT NL NO • RELIENE, Ramune PL PT RO RS SE SI SK SM TR Los Angeles, CA California 90024 (US) • BORNEMAN, James (30) Priority: 22.02.2013 US 201361956186 P Riverside, CA California 92506 (US) 26.11.2013 US 201361909242 P • PRESLEY, Laura, L. Santa Maria, CA California 93458 (US) (62) Document number(s) of the earlier application(s) in • BRAUN, Jonathan accordance with Art. 76 EPC: Tarzana, CA California 91356 (US) 14753847.4 / 2 958 575 (74) Representative: Müller-Boré & Partner (71) Applicant: The Regents of the University of Patentanwälte PartG mbB California Friedenheimer Brücke 21 Oakland, CA 94607 (US) 80639 München (DE) (72) Inventors: Remarks: • YAMAMOTO, Mitsuko, L. This application was filed on 27-03-2019 as a Alameda, CA California 94502 (US) divisional application to the application mentioned under INID code 62. (54) FOOD OR BEVERAGE PRODUCT, OR PROBIOTIC COMPOSITION, COMPRISING LACTOBACILLUS JOHNSONII 456 (57) The present invention relates to food products, beverage products and probiotic compositions comprising Lacto- bacillus johnsonii 456. EP 3 536 328 A1 Printed by Jouve, 75001 PARIS (FR) EP 3 536 328 A1 Description CROSS-REFERENCE TO RELATED APPLICATIONS 5 [0001] This application claims the benefit of U.S. -

Genomics of Helicobacter Species 91
Genomics of Helicobacter Species 91 6 Genomics of Helicobacter Species Zhongming Ge and David B. Schauer Summary Helicobacter pylori was the first bacterial species to have the genome of two independent strains completely sequenced. Infection with this pathogen, which may be the most frequent bacterial infec- tion of humanity, causes peptic ulcer disease and gastric cancer. Other Helicobacter species are emerging as causes of infection, inflammation, and cancer in the intestine, liver, and biliary tract, although the true prevalence of these enterohepatic Helicobacter species in humans is not yet known. The murine pathogen Helicobacter hepaticus was the first enterohepatic Helicobacter species to have its genome completely sequenced. Here, we consider functional genomics of the genus Helico- bacter, the comparative genomics of the genus Helicobacter, and the related genera Campylobacter and Wolinella. Key Words: Cytotoxin-associated gene; H-Proteobacteria; gastric cancer; genomic evolution; genomic island; hepatobiliary; peptic ulcer disease; type IV secretion system. 1. Introduction The genus Helicobacter belongs to the family Helicobacteriaceae, order Campylo- bacterales, and class H-Proteobacteria, which is also known as the H subdivision of the phylum Proteobacteria. The H-Proteobacteria comprise of a relatively small and recently recognized line of descent within this extremely large and phenotypically diverse phy- lum. Other genera that colonize and/or infect humans and animals include Campylobac- ter, Arcobacter, and Wolinella. These organisms are all microaerophilic, chemoorgano- trophic, nonsaccharolytic, spiral shaped or curved, and motile with a corkscrew-like motion by means of polar flagella. Increasingly, free living H-Proteobacteria are being recognized in a wide range of environmental niches, including seawater, marine sedi- ments, deep-sea hydrothermal vents, and even as symbionts of shrimp and tubeworms in these environments. -

Co-Infection Associated with Diarrhea in a Colony of <I>Scid
Laboratory Animal Science Vol 48, No 5 Copyright 1998 October 1998 by the American Association for Laboratory Animal Science Helicobacter bilis/Helicobacter rodentium Co-Infection Associated with Diarrhea in a Colony of scid Mice Nirah H. Shomer,* Charles A. Dangler, Robert P. Marini, and James G. Fox† Abstract _ An outbreak of diarrhea spanning 3 months occurred in a breeding colony of scid/Trp53 knockout mice. Approximately a third of the 150 mice were clinically affected, with signs ranging from mucoid or watery diarrhea to severe hemorrhagic diarrhea with mortality. Helicobacter bilis and the newly recognized urease-negative organ- ism H. rodentium were isolated from microaerobic culture of feces or cecal specimens from affected mice. Dual infection with H. bilis and H. rodentium were confirmed by culture and polymerase chain reaction (PCR) in several animals. Both Helicobacter species rapidly colonized immunocompetent sentinel mice exposed to bedding from cages containing affected mice, but the sentinel remained asymptomatic. Mice with diarrhea had multifocal to segmental proliferative typhlitis, colitis, and proctitis. Several affected mice had multifocal mucosal necrosis with a few focal ulcers in the cecum, colon, and rectum. Mice with diarrhea were treated with antibiotic food wafers (1.5 mg of amoxicillin, 0.69 mg of metronidazole, and 0.185 mg of bismuth/mouse per day) previously shown to eradi- cate H. hepaticus in immunocompetent mice. Antibiotic treatment resulted in resolution of diarrhea, but not eradication of H. bilis and H. rodentium; mice continued to have positive PCR results after a 2-week treatment regimen, and clinical signs of diarrhea returned in some mice when treatment was suspended. -

Diagnostic Assay for Helicobacter Hepaticus Based on Nucleotide Sequence of Its 16S Rrna Gene JANE K
JOURNAL OF CLINICAL MICROBIOLOGY, May 1995, p. 1344–1347 Vol. 33, No. 5 0095-1137/95/$04.0010 Copyright q 1995, American Society for Microbiology Diagnostic Assay for Helicobacter hepaticus Based on Nucleotide Sequence of Its 16S rRNA Gene JANE K. BATTLES,1 JAMES C. WILLIAMSON,1 KRISTEN M. PIKE,1 PETER L. GORELICK,2 3 1 JERROLD M. WARD, AND MATTHEW A. GONDA * Laboratory of Cell and Molecular Structure1 and Laboratory Animal Sciences Program,2 Program Resources, Inc./DynCorp, and Veterinary and Tumor Pathology Section, Office of Laboratory Animal Science,3 National Cancer Institute-Frederick Cancer Research and Development Center, Frederick, Maryland 21702-1201 Received 17 November 1994/Returned for modification 3 January 1995/Accepted 7 February 1995 Conserved primers were used to PCR amplify 95% of the Helicobacter hepaticus 16S rRNA gene. Its sequence was determined and aligned to those of related bacteria, enabling the selection of primers to highly diverged regions of the 16S rRNA gene and an oligonucleotide probe for the development of a PCR-liquid hybridization assay. This assay was shown to be both sensitive and specific for H. hepaticus 16S rRNA gene sequences. Helicobacter hepaticus is a recently identified species of Helicobacter canis (34). Many PCR-based techniques have gram-negative, microaerophilic, urease-positive, spiral bacte- been developed to amplify 16S rRNA sequences of H. pylori rium that was originally isolated from the livers of mice with and related organisms (3, 6, 15, 16, 20, 24, 39, 44). chronic active hepatitis at the National Cancer Institute-Fred- In the present report, the objective was to develop a species- erick Cancer Research and Development Center. -

The Molecular Phylogeny and Ecology of Spiral Bacteria from the Mouse Gastrointestinal Tract
The Molecular Phylogeny and Ecology of Spiral Bacteria from the Mouse Gastrointestinal Tract Bronwyn Ruth Robertson A thesis submitted for the degree of Doctor of Philosophy School of Microbiology and Immunology The University of New South Wales Sydney, Australia May, 1998 'Brief rejfection on test-tu.ies 'Ta~ a piece offire, a piece ofwater, a piece of ra66it or a piece of tree, or any piece ofa liuman 6eing, ~ it, slia~ it, stopper it up, k.._eep it wann, in tlie tfarl<:.i in tlie Bglit, refrigerate/, fet it stantf stifffor a wliife - yourselves far from stiff- 6ut that's tlie realjo~. Jtjter a wliife you wok.._- ~ntf it's growing, a fittfe ocean, a fittle vofcano, a fittfe tree, a fittfe lieart, a fittfe 6rain, so fittfe you don't liear it lamenting as it wants to get out, 6ut that's tlie reafjo~, not liearing it. 'Ift.engo ·antf record it, a[[ tfaslies or a[[ crosses, some witli ~famation-mar/&, a[[ nouglits antf a[[figures, some witli ~famation-marf&, antf that's tlie reafjo~, in effect a test-tu6e is a device for changing nouglits into ~famation mar/&. 'Iliat's tlie reafJo~ wliicli mak.._es you forget for a wliile tliat reaffy you yourself are In tlie test-tu6e Mirosfav !Jfo{u6 Poems 'Before arufJtfter Acknowledgements I extend my grateful thanks to the following people for their assistance and encouragement during my PhD studies. Professor Adrian Lee for giving me the opportunity to carry out my PhD in his laboratory, for his supervision and for his enthusiasm for the "other helicobacters". -

List of the Pathogens Intended to Be Controlled Under Section 18 B.E
(Unofficial Translation) NOTIFICATION OF THE MINISTRY OF PUBLIC HEALTH RE: LIST OF THE PATHOGENS INTENDED TO BE CONTROLLED UNDER SECTION 18 B.E. 2561 (2018) By virtue of the provision pursuant to Section 5 paragraph one, Section 6 (1) and Section 18 of Pathogens and Animal Toxins Act, B.E. 2558 (2015), the Minister of Public Health, with the advice of the Pathogens and Animal Toxins Committee, has therefore issued this notification as follows: Clause 1 This notification is called “Notification of the Ministry of Public Health Re: list of the pathogens intended to be controlled under Section 18, B.E. 2561 (2018).” Clause 2 This Notification shall come into force as from the following date of its publication in the Government Gazette. Clause 3 The Notification of Ministry of Public Health Re: list of the pathogens intended to be controlled under Section 18, B.E. 2560 (2017) shall be cancelled. Clause 4 Define the pathogens codes and such codes shall have the following sequences: (1) English alphabets that used for indicating the type of pathogens are as follows: B stands for Bacteria F stands for fungus V stands for Virus P stands for Parasites T stands for Biological substances that are not Prion R stands for Prion (2) Pathogen risk group (3) Number indicating the sequence of each type of pathogens Clause 5 Pathogens intended to be controlled under Section 18, shall proceed as follows: (1) In the case of being the pathogens that are utilized and subjected to other law, such law shall be complied. (2) Apart from (1), the law on pathogens and animal toxin shall be complied. -

Genomic Analysis of Helicobacter Himalayensis Sp. Nov. Isolated from Marmota Himalayana
Genomic analysis of Helicobacter himalayensis sp. nov. isolated from Marmota himalayana Shouhui Hu Peking University Shougang Hospital Lina Niu Hainan Medical University Lei Wu Peking University Shougang Hospital Xiaoxue Zhu Peking University Shougang Hospital Yu Cai Peking University Shougang Hospital Dong Jin Chinese Center for Disease Control and Prevention Linlin Yan Peking University Shougang Hospital Fan Zhao ( [email protected] ) Peking University Shougang Hospital https://orcid.org/0000-0002-8164-5016 Research article Keywords: Helicobacter, Comparative genomics, Helicobacter himalayensis, Virulence factor Posted Date: December 1st, 2020 DOI: https://doi.org/10.21203/rs.3.rs-55448/v3 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on November 23rd, 2020. See the published version at https://doi.org/10.1186/s12864-020-07245-y. Page 1/18 Abstract Background: Helicobacter himalayensis was isolated from Marmota himalayana in the Qinghai-Tibet Plateau, China, and is a new non-H. pylori species, with unclear taxonomy, phylogeny, and pathogenicity. Results: A comparative genomic analysis was performed between the H. himalayensis type strain 80(YS1)T and other the genomes of Helicobacter species present in the National Center for Biotechnology Information (NCBI) database to explore the molecular evolution and potential pathogenicity of H. himalayensis. H. himalayensis 80(YS1)T formed a clade with H. cinaedi and H. hepaticus that was phylogenetically distant from H. pylori. The H. himalayensis genome showed extensive collinearity with H. hepaticus and H. cinaedi. However, it also revealed a low degree of genome collinearity with H. -
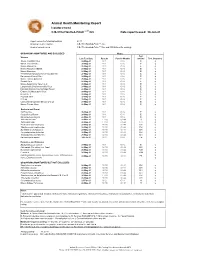
2021 ARC Health Report
Animal Health Monitoring Report Isolator reared C.B-17/IcrHanHsd-Prkdc scid /Arc Date report issued: 30-Jun-21 Report covers the following isolators: RI 27 Strains present in isolator: C.B-17/IcrHanHsd-Prkdc scid /Arc Strain of animal tested: C.B-17/IcrHanHsd-Prkdc scid /Arc and ICR Outbred for serology ORGANISMS MONITORED AND EXCLUDED Mouse Test Viruses Last Test Date Results Past 18 Months method Test frequency Mouse Hepatitis Virus 26-May-21 0 / 1 0 / 6 E q Minute Virus of Mice 26-May-21 0 / 1 0 / 6 E q Mouse Parvovirus 26-May-21 0 / 1 0 / 6 E q Murine Rotavirus (EDIM) 26-May-21 0 / 1 0 / 6 E q Mouse Norovirus 26-May-21 0 / 1 0 / 6 E q Theiler's Encephalomyelitis Virus (GD VII) 26-May-21 0 / 1 0 / 6 E q Pneumonia Virus of Mice 26-May-21 0 / 1 0 / 6 E q Murine Cytomegalovirus 26-May-21 0 / 1 0 / 6 E q Sendai Virus 26-May-21 0 / 1 0 / 6 E q Mouse Adenovirus Type 1 & 2 26-May-21 0 / 1 0 / 6 E q Lymphocytic Choriomeningitis Virus 26-May-21 0 / 1 0 / 6 E q Hantaan (Korean Haemorrhagic Fever) 26-May-21 0 / 1 0 / 6 E q Ectromelia (Mousepox) Virus 26-May-21 0 / 1 0 / 6 E q Reovirus -3 26-May-21 0 / 1 0 / 6 E q Polyoma Virus 26-May-21 0 / 1 0 / 6 E q K Virus 26-May-21 0 / 1 0 / 6 E q Lactic Dehydrogenase Elevating Virus 26-May-21 0 / 1 0 / 6 E q Mouse Thymic Virus 26-May-21 0 / 1 0 / 6 E q 00-Jan-00 Bacteria and Fungi CAR bacillus 26-May-21 0 / 1 0 / 6 E q Clostridium piliforme 26-May-21 0 / 1 0 / 6 E q Mycoplasma pulmonis 26-May-21 0 / 1 0 / 6 E q Helicobacter spp.1 26-May-21 0 / 10 0 / 44 H q Salmonella spp. -
R Graphics Output
883 | Desulfovibrio vulgaris | DvMF_2825 298701 | Desulfovibrio | DA2_3337 1121434 | Halodesulfovibrio aestuarii | AULY01000007_gene1045 207559 | Desulfovibrio alaskensis | Dde_0991 935942 | Desulfonatronum lacustre | KI912608_gene2193 159290 | Desulfonatronum | JPIK01000018_gene1259 1121448 | Desulfovibrio gigas | DGI_0655 1121445 | Desulfovibrio desulfuricans | ATUZ01000018_gene2316 525146 | Desulfovibrio desulfuricans | Ddes_0159 665942 | Desulfovibrio | HMPREF1022_02168 457398 | Desulfovibrio | HMPREF0326_00453 363253 | Lawsonia intracellularis | LI0397 882 | Desulfovibrio vulgaris | DVU_0784 1121413 | Desulfonatronovibrio hydrogenovorans | JMKT01000008_gene1463 555779 | Desulfonatronospira thiodismutans | Dthio_PD0935 690850 | Desulfovibrio africanus | Desaf_1578 643562 | Pseudodesulfovibrio aespoeensis | Daes_3115 1322246 | Pseudodesulfovibrio piezophilus | BN4_12523 641491 | Desulfovibrio desulfuricans | DND132_2573 1121440 | Desulfovibrio aminophilus | AUMA01000002_gene2198 1121456 | Desulfovibrio longus | ATVA01000018_gene290 526222 | Desulfovibrio salexigens | Desal_3460 1121451 | Desulfovibrio hydrothermalis | DESAM_21057 1121447 | Desulfovibrio frigidus | JONL01000008_gene3531 1121441 | Desulfovibrio bastinii | AUCX01000006_gene918 1121439 | Desulfovibrio alkalitolerans | dsat_0220 941449 | Desulfovibrio | dsx2_0067 1307759 | Desulfovibrio | JOMJ01000003_gene2163 1121406 | Desulfocurvus vexinensis | JAEX01000012_gene687 1304872 | Desulfovibrio magneticus | JAGC01000003_gene2904 573370 | Desulfovibrio magneticus | DMR_04750 -

Novel Mechanisms and Therapies for Celiac Disease
NOVEL MECHANISMS AND THERAPIES FOR CELIAC DISEASE EXPLORING NOVEL MECHANISMS AND THERAPIES FOR CELIAC DISEASE By HEATHER J GALIPEAU, B.H.Sc A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by Heather J Galipeau, 2015 PhD Thesis- H.J. Galipeau; McMaster University- Medical Sciences DESCRIPTIVE NOTE Doctor of Philosophy (2015) McMaster University, Hamilton, Ontario (Medical Sciences) TITLE Exploring novel mechanisms and therapies for celiac disease AUTHOR Heather J. Galipeau, B.H.Sc SUPERVISOR Dr. Elena F. Verdu NUMBER OF PAGES xix, 314 ii PhD Thesis- H.J. Galipeau; McMaster University- Medical Sciences ABSTRACT The gastrointestinal tract forms the body’s largest interface with the external environment and is exposed to a vast amount of foreign material, including pathogenic and commensal bacteria, as well as food antigens. The gastrointestinal tract has multiple functions that are performed through complex interactions by its different components. It must be able to degrade food, absorb nutrients and eliminate waste, while at the same time maintain a balance between immune tolerance and protection against pathogenic and antigenic material. This concept of mucosally-induced tolerance is a key feature of the gut immune system, whereby a state of local and systemic unresponsiveness to food protein or systemic ignorance of commensal bacteria is maintained under homeostatic conditions through interactions between the host, dietary factors, and the intestinal microbiota. Dysfunctional interactions can lead to a breakdown in tolerance to otherwise innocuous antigens. One of the best characterized food sensitivities is celiac disease (CD). -

The Following Pages Constitute the Final, Accepted and Revised Manuscript of the Article
The following pages constitute the final, accepted and revised manuscript of the article: Nilsson, Hans-Olof and Pietroiusti, Antonio and Gabrielli, Maurizio and Zocco, Maria Assunta and Gasbarrini, Giovanni and Gasbarrini, Antonio “Helicobacter pylori and Extragastric Diseases - Other Helicobacters” Helicobacter. 2005;10 Suppl 1:54-65. Publisher: Blackwell Use of alternative location to go to the published version of the article requires journal subscription. Alternative location: http://dx.doi.org/10.1111/j.1523-5378.2005.00334.x HELICOBACTER PYLORI AND EXTRAGASTRIC DISEASES - OTHER HELICOBACTERS Hans-Olof Nilsson, Antonio Pietroiusti*, Maurizio Gabrielli#, Maria Assunta Zocco#, Giovanni Gasbarrini#, Antonio Gasbarrini# Department of Laboratory Medicine, Lund University, Lund, Sweden *Medical Semiology and Methodology, Department of Internal Medicine, Tor Vergata University, Rome, Italy #Department of Internal Medicine, Catholic University the Sacred Heart, Gemelli Hospital Rome, Italy Correspondence and reprints request to: Antonio Gasbarrini, MD Istituto di Patologia Speciale Medica Universita’ Cattolica del Sacro Cuore Policlinico Gemelli, Largo Gemelli 8, 00168 Rome, ITALY Telephone: 39-335-6873562 39-6-30154294 FAX: 39-6-35502775 E-mail: [email protected] 2 ABSTRACT The involvement of Helicobacter pylori in the pathogenesis of extragastric diseases continues to be an interesting topic in the field of Helicobacter-related pathology. Although conflicting findings have been reported for most of the disorders, a role of H. pylori seems to be important especially for the development of cardiovascular and hematologic disorders. Previously isolated human and animal Helicobacter sp. flexispira and ′Helicobacter heilmannii′ strains have been validated using polyphasic taxonomy. A novel enterohepatic helicobacter has been isolated from mastomys and mice, adding to the list of helicobacters that colonize the liver. -

Modulation of Intestinal Homeostasis and Inflammation by Prevotella Intestinalis (Nov
Modulation of intestinal homeostasis and inflammation by Prevotella intestinalis (nov. sp.) Von der Fakultät für Lebenswissenschaften der Technischen Universität Carolo-Wilhelmina zu Braunschweig zur Erlangung des Grades einer Doktorin der Naturwissenschaften (Dr. rer. nat.) genehmigte D i s s e r t a t i o n von Aida Iljazovic aus Rijeka, Kroatien 1. Referentin: Prof. Dr. Petra Dersch 2. Referent: Prof. Dr. Stefan Dübel eingereicht am: 25.07.2018 mündliche Prüfung (Disputation) am: 26.10.2018 Druckjahr 2019 Vorveröffentlichungen der Dissertation Teilergebnisse aus dieser Arbeit wurden mit Genehmigung der Fakultät für Lebenswissenschaften, vertreten durch die Mentorin der Arbeit, in folgenden Beiträgen vorab veröffentlicht: Tagungsbeiträge Aida Iljazovic, Eric J.C. Galvez, Till-Robin Lesker, Sophie Thiemann and Till Strowig: Microbial Interactions of Prevotella spp. within the Intestinal Ecosystem. Presentation. 9th Seeon Conference on „Microbiota, Probiota and Host“, 24-26 June 2016, Kloster Seeon, Germany. Aida Iljazovic, Eric J.C. Galvez, Till-Robin Lesker, Sophie Thiemann and Till Strowig: Microbial Interactions of Prevotella spp. within the Intestinal Ecosystem. Poster. Symposium Microbiota and Mucosal Immunity: Rules of Engagement in Health and Disease, 28-30 July 2016. Toronto, Canada. Per Marco, senza il cui amore e supporto la fine di questa dissertazione sarebbe stata impensabile. Acknowledgments Doing a PhD research often appears a solitary undertaking. However, it is unthinkable to maintain the degree of dedication and focus necessary for its completion without the help and support of many people. I take this opportunity to extend my sincere gratitude and appreciation to all those who made this PhD thesis possible. Foremost I would like to thank my supervisor, Dr.