A Fully Featured COMBINE Archive of a Simulation Study
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biological Pathways Exchange Language Level 3, Release Version 1 Documentation
BioPAX – Biological Pathways Exchange Language Level 3, Release Version 1 Documentation BioPAX Release, July 2010. The BioPAX data exchange format is the joint work of the BioPAX workgroup and Level 3 builds on the work of Level 2 and Level 1. BioPAX Level 3 input from: Mirit Aladjem, Ozgun Babur, Gary D. Bader, Michael Blinov, Burk Braun, Michelle Carrillo, Michael P. Cary, Kei-Hoi Cheung, Julio Collado-Vides, Dan Corwin, Emek Demir, Peter D'Eustachio, Ken Fukuda, Marc Gillespie, Li Gong, Gopal Gopinathrao, Nan Guo, Peter Hornbeck, Michael Hucka, Olivier Hubaut, Geeta Joshi- Tope, Peter Karp, Shiva Krupa, Christian Lemer, Joanne Luciano, Irma Martinez-Flores, Zheng Li, David Merberg, Huaiyu Mi, Ion Moraru, Nicolas Le Novere, Elgar Pichler, Suzanne Paley, Monica Penaloza- Spinola, Victoria Petri, Elgar Pichler, Alex Pico, Harsha Rajasimha, Ranjani Ramakrishnan, Dean Ravenscroft, Jonathan Rees, Liya Ren, Oliver Ruebenacker, Alan Ruttenberg, Matthias Samwald, Chris Sander, Frank Schacherer, Carl Schaefer, James Schaff, Nigam Shah, Andrea Splendiani, Paul Thomas, Imre Vastrik, Ryan Whaley, Edgar Wingender, Guanming Wu, Jeremy Zucker BioPAX Level 2 input from: Mirit Aladjem, Gary D. Bader, Ewan Birney, Michael P. Cary, Dan Corwin, Kam Dahlquist, Emek Demir, Peter D'Eustachio, Ken Fukuda, Frank Gibbons, Marc Gillespie, Michael Hucka, Geeta Joshi-Tope, David Kane, Peter Karp, Christian Lemer, Joanne Luciano, Elgar Pichler, Eric Neumann, Suzanne Paley, Harsha Rajasimha, Jonathan Rees, Alan Ruttenberg, Andrey Rzhetsky, Chris Sander, Frank Schacherer, -

SBML Level 3: an Extensible Format for the Exchange and Reuse of Biological Models
Review SBML Level 3: an extensible format for the exchange and reuse of biological models Sarah M Keating1,2,3,† , Dagmar Waltemath4,† , Matthias König5 , Fengkai Zhang6 , Andreas Dräger7,8,9 , Claudine Chaouiya10,11 , Frank T Bergmann3 , Andrew Finney12, Colin S Gillespie13 , Tomás Helikar14 , Stefan Hoops15 , Rahuman S Malik-Sheriff2 , Stuart L Moodie16 , Ion I Moraru17 , Chris J Myers18 , Aurélien Naldi19 , Brett G Olivier1,3,20 , Sven Sahle3, James C Schaff21 , Lucian P Smith1,22 , Maciej J Swat23, Denis Thieffry19 , Leandro Watanabe18 , Darren J Wilkinson13,24 , Michael L Blinov17 , Kimberly Begley25 , James R Faeder26 , Harold F Gómez27, Thomas M Hamm7,8 , Yuichiro Inagaki28 , Wolfram Liebermeister29 , Allyson L Lister30 , Daniel Lucio31 , Eric Mjolsness32 , Carole J Proctor33 , Karthik Raman34,35,36 , Nicolas Rodriguez37 , Clifford A Shaffer38 , Bruce E Shapiro39, Joerg Stelling40 , Neil Swainston41 , Naoki Tanimura42, John Wagner43, Martin Meier-Schellersheim6 , Herbert M Sauro22 , Bernhard Palsson44 , Hamid Bolouri45, Hiroaki Kitano46,47 , Akira Funahashi48 , Henning Hermjakob2 , John C Doyle1 , Michael Hucka1,* & SBML Level 3 Community members‡ Abstract ways of integrating data with models. We provide our perspectives on the challenges presented by these developments and how SBML Systems biology has experienced dramatic growth in the number, size, Level 3 provides the foundation needed to support this evolution. and complexity of computational models. To reproduce simulation results and reuse models, researchers must exchange unambiguous Keywords computational modeling; file format; interoperability; reproducibil- model descriptions. We review the latest edition of the Systems Biol- ity; systems biology ogy Markup Language (SBML), a format designed for this purpose. A Subject Categories Computational Biology; Metabolism; Methods and community of modelers and software authors developed SBML Level Resources 3 over the past decade. -

Specifications of Standards in Systems and Synthetic Biology
Journal of Integrative Bioinformatics, 12(2):258, 2015 http://journal.imbio.de/ Specifications of Standards in Systems and Synthetic Biology Falk Schreiber1*, Gary D. Bader2, Martin Golebiewski3, Michael Hucka4, Benjamin Kormeier5, Nicolas Le Novere` 6, Chris Myers7, David Nickerson8, Bjorn¨ Sommer9, Dagmar Waltemath10 and Stephan Weise11 1Faculty of IT, Monash University, Clayton, Australia & Martin Luther University Halle-Wittenberg, Germany 2The Donnelly Centre, University of Toronto, Canada 3Heidelberg Institute for Theoretical Studies (HITS), Germany 4California Institute of Technology, USA 5University of Bielefeld, Germany 6Babraham Institute, UK 7University of Utah, USA 8Auckland Bioengineering Institute, University of Auckland, New Zealand 9Faculty of IT, Monash University, Clayton, Australia 10University of Rostock, Germany 11Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany Standards shape our everyday life. From nuts and bolts to electronic devices and technological processes, standardised products and processes are all around us. Standards have technological and economic benefits, such as making information exchange, production, and services more efficient. However, novel, innovative areas often either lack proper standards, or documents about standards in these areas are not available from a centralised platform or formal body (such as the International Standardisation Organisation). Systems and synthetic biology is a relatively novel area, and it is only in the last decade that the standardisation of data, information, and models related to systems and synthetic biology Copyright 2015 The Author(s). PublishedThis by article Journal is of licensed Integrative under Bioinformatics. a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License (http://creativecommons.org/licenses/by-nc-nd/3.0/). has become a community-wide effort. -
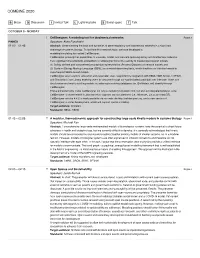
Combine 2020
COMBINE 2020 B Break D Discussion I Invited Talk L Lightning talks S Social space T Talk OCTOBER 5 • MONDAY I CellDesigner: A modeling tool for biochemical networks Room 1 PINNED Speakers: Akira Funahashi 01:00 – 01:45 Abstract: Understanding the logic and dynamics of gene-regulatory and biochemical networks is a significant challenge of systems biology. To facilitate this research topic, we have developed a modeling/simulating tool called CellDesigner. CellDesigner primarily has capabilities to visualize, model, and simulate gene-regulatory and biochemical networks. Two significant characteristics embedded in CellDesigner boost its usability to create/import/export models: (1) Solidly defined and comprehensive graphical representation (Process Diagram) of network models and (2) Systems Biology Markup Language (SBML) as a model-describing basis, which functions as inter-tool media to import/export SBML-based models. CellDesigner also supports simulation and parameter scan, supported by integration with SBML ODE Solver, COPASI, and Simulation Core Library enabling users to simulate through our sophisticated graphical user interface. Users can also browse and modify existing models by referring to existing databases (ex. BioModels.net) directly through CellDesigner. These enhancements make CellDesigner not only a modeling/simulation tool, but also an integrated analysis suite. CellDesigner is implemented in Java and thus supports various platforms (i.e., Windows, Linux, and macOS). CellDesigner version 4.4.2 is freely available via our web site http://celldesigner.org, and a new version of CellDesigner is under development, which will support spatial modeling. Target audience: modelers Standards: SBML, SBGN 01:45 – 02:00 T A modular, thermodynamic approach for constructing large-scale kinetic models in systems biology Room 1 Speakers: Michael Pan Abstract: Comprehensive large-scale mathematical models of biomolecular systems have the potential to direct future advances in health and biotechnology, but are currently difficult to develop. -
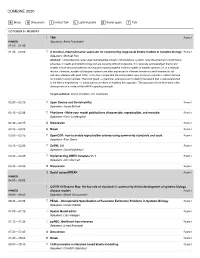
Combine 2020
COMBINE 2020 B Break D Discussion I Invited Talk L Lightning talks S Social space T Talk OCTOBER 5 • MONDAY I TBA Room 1 PINNED Speakers: Akira Funahashi 01:00 – 01:45 01:45 – 02:00 T A modular, thermodynamic approach for constructing large-scale kinetic models in systems biology Room 1 Speakers: Michael Pan Abstract: Comprehensive large-scale mathematical models of biomolecular systems have the potential to direct future advances in health and biotechnology, but are currently difficult to develop. It is generally acknowledged that kinetic models should be constructed by reusing and coupling together existing models of smaller systems, i.e. in a modular fashion. However, models of biological systems are often expressed in different formalisms and therefore do not naturally interface with each other. In this talk, I argue that the conservation laws of physics provide a unified interface for models to communicate. The bond graph - a graphical, energy-based modelling framework that is well-established in the field of engineering - is introduced as a means of enabling this approach. The approach will be illustrated in the development of a model of the MAPK signalling cascade. Target audience: kinetic modellers, tool developers 02:00 – 02:15 T Open Source and Sustainability Room 1 Speakers: Jacob Barhak 02:15 – 02:30 T Physiome - Make your model publications discoverable, reproducible, and reusable. Room 1 Speakers: Karin Lundengård 02:30 – 02:45 D Discussion Room 1 02:45 – 03:00 B Break Room 1 03:00 – 03:15 T OpenCOR: how to enable reproducible -

Interoperable Standards for Modelling in Biology
Interoperable Standards For modelling in biology Rising interest in models from new stakeholders ● “Biologists”: computational models look “useful”, “serious” ● Publishers: computational articles are respectable, can be published in high profile journals ● Funding agencies: Models could help with the major challenges (read “science that can be sold to citizen/electors”): Health, Food, Energy... ● Industries: Models could help with the major challenges (read “new opportunities to make money”): Pharmas, crops, biofuels ... Computational models on the rise BioModels Database (published models branch) growth since its creation http://www.ebi.ac.uk/biomodels/ We need to share: ModelModel descriptionsdescriptions SimulationSimulation descriptionsdescriptions ParametrisationsParametrisations In order to: Re-useRe-use VerifyVerify ModifyModify BuildBuild uponupon IntegrateIntegrate withwith What are standards good for (1)? What are standards good for (1)? N tools require N conversions for exchange and not N(N-1)/2 What are standards good for (2)? Open standards are more stable than proprietary formats What are standards good for (3)? SBToolbox2 MatLab SBMLToolbox CySBML Cytoscape libSBML JSBML SBML Open standards can be built on. They generate new science Many alternative modelling approaches State-Transitions, cable Neurobiology Approximation (PDE) Physiology Biochemistry Process Variable description Descriptions (ODE, PDE) (ODE, Monte-Carlo) Rule-based models Qualitative models Cell automata Multi-agents, Matrix models Developmental biology, -

Physiome Model Repository.Pptx
The Physiome Model Repository Poul Nielsen • The Physiome model repository (PMR) is a freely accessible site for researchers to find, store, and share models. • Contains over 500 published models encoded in CellML, and some examples of FieldML models. The PHYSIOME model repository • NOT the CellML model repository. • NOT the FieldML model repository. • IS a useful repository designed to improve collaboration in model development and dissemination. • Use of Physiome/COMBINE standards (BioPAX, CellML, SBGN, SBML, SED-ML) enhances collaboration and dissemination… • …but adherence is not required in order to put your work in the repository! Workspace • All models exist in workspaces. • A workspace is the basic unit for collecting one or more related files. • Workspaces can store any kind of file: • CellML; SBML; FieldML; SED-ML; PDF; .doc; … • .m; .c; .cpp; .f; .f90; .py; ... • .com; .ipnode; .ipelem; .ipequa; .ipgrid; .ipcell; … • Not restricted to models: • experimental data; • simulation results; • images; • journal publications; • … Viewing list of workspaces Viewing a particular workspace Workspace provenance • Workspaces are version controlled. • Every revision to a workspace is recorded as a changeset. • A changeset: • is an immutable representation of the contents of the workspace. • is a complete and unambiguous record of the workspace evolution; • has a unique URL for citation; • is linked to authors responsible for changes. Viewing changesets in a workspace Workspace management • Workspaces are managed. • All workspaces have access control: • who can read from and write to a given workspace; • controls the visibility of workspaces to nominated individuals and/or groups. • Interface workflows enable: • management of state transitions (e.g. private → public → published); • oversight and control by repository curators. -

Journal of Integrative Bioinformatics 2020; 20200022
Journal of Integrative Bioinformatics 2020; 20200022 Editorial Falk Schreiber*, Björn Sommer, Tobias Czauderna, Martin Golebiewski, Thomas E. Gorochowski, Michael Hucka, Sarah M. Keating, Matthias König, Chris Myers, David Nickerson and Dagmar Waltemath Specifications of standards in systems and synthetic biology: status and developments in 2020 https://doi.org/10.1515/jib-2020-0022 Received April 26, 2020; accepted April 27, 2020 Abstract: This special issue of the Journal of Integrative Bioinformatics presents papers related to the 10th COMBINE meeting together with the annual update of COMBINE standards in systems and synthetic biology. Keywords: ontologies; standards; systems biology; synthetic biology. 1 Introduction COMBINE (‘COmputational Modelling in BIology’ NEtwork) [1, 2], the formal entity which coordinates the development of standards in systems and synthetic biology, celebrated 10 years of activity in 2019. COMBINE not only coordinates standard developments, but also fosters and moderates discussions; designs and implements dissemination strategies; and organises two annual community meetings – COMBINE and HARMONY. This special issue contains two papers, one new standard, and six updates of standards. Waltemath et al. [3] discuss the first 10 years of the international coordination network for standards in systems and synthetic biology, and summarises the COMBINE meeting in Heidelberg in July 2019. Brunak et al. [4] present ongoing works and open questions towards standardisation guidelines for in silico approaches in personalised medicine. COMBINE standards and associated initiatives cover a wide range of disciplines, see Figure 1. This special issue only highlights updates over the last year, namely the CellML 2.0 specification, the Systems Biology Graphical Notation - Markup Language (SBGN-ML) Milestone 3, the SBML (Systems Biology Markup Language) Level 3 Packages “Distrib” and “Multi”, the SBML (Systems Biology Markup Language) Version 3.0.0, and the SBOL (Synthetic Biology Open Language) Visual Version 2.2. -

A Brief History of Combine
Proceedings of the 2017 Winter Simulation Conference W. K. V. Chan, A. D’Ambrogio, G. Zacharewicz, N. Mustafee, G. Wainer, and E. Page, eds. A BRIEF HISTORY OF COMBINE Chris J. Myers Gary Bader University of Utah University of Toronto 50 S. Central Campus Dr., Rm. 2110 160 College St., Rm. 602 Salt Lake City, UT 84112, USA Toronto, ON M5S 3E1, CANADA Padraig Gleeson Martin Golebiewski University College London HITS gGmbH London - WC1E 6BT Schloss-Wolfsbrunnenweg 35 UNITED KINGDOM 69118 Heidelberg, GERMANY Michael Hucka Nicolas Le Novere` California Institute of Technology The Babraham Institute 1200 E. California Blvd. Cambridge CB22 3LF Pasadena, CA 91125, USA UNITED KINGDOM David P. Nickerson Falk Schreiber University of Auckland University of Konstanz 70 Symonds Street Universitatsstr.¨ 10 Auckland, NEW ZEALAND 78464 Konstanz, GERMANY Dagmar Waltemath University of Rostock Albert-Einstein-Str. 22 18059 Rostock, GERMANY ABSTRACT Standards for data exchange are critical to the development of any field. They enable researchers and practitioners to transport information reliably, to apply a variety of tools to their problems, and to reproduce scientific results. Over the past two decades, a range of standards have been developed to facilitate the exchange and reuse of information in the domain of representation and modeling of biological systems. These standards are complementary, so the interactions between their developers increased over time. By the end of the last decade, the community of researchers decided that more interoperability is required between the standards, and that common development is needed to make better use of effort, time, and money devoted to this activity. -

The Biopax Community Standard for Pathway Data Sharing
Biology Faculty Works Biology 9-2010 The BioPAX Community Standard for Pathway Data Sharing E. Demir M. P. Cary S. Paley K. Fukuda C. Lemer See next page for additional authors Follow this and additional works at: https://digitalcommons.lmu.edu/bio_fac Part of the Biology Commons Recommended Citation Dahlquist, Kam [et al]. 2010."The BioPAX Community Standard for Pathway Data Sharing." Nature Biotechnology 28 (9): 935-42. This Article is brought to you for free and open access by the Biology at Digital Commons @ Loyola Marymount University and Loyola Law School. It has been accepted for inclusion in Biology Faculty Works by an authorized administrator of Digital Commons@Loyola Marymount University and Loyola Law School. For more information, please contact [email protected]. Authors E. Demir, M. P. Cary, S. Paley, K. Fukuda, C. Lemer, I. Vastrik, G. Wu, P. D’Eustachio, C. Schaefer, J. Luciano, F. Schacherer, I. Martinez-Flores, Z. Hu, V. Jimenez-Jacinto, G. Joshi-Tope, K. Kandasamy, A. C. Lopez- Fuentes, H. Mi, E. Pichler, I. Rodchenkov, A. Splendiani, S. Tkachev, J. Zucker, G. Gopinath, H. Rajasimha, R. Ramakrishnan, I. Shah, M. Syed, N. Anwar, O. Babur, M. Blinov, E. Brauner, D. Corwin, S. Donaldson, F. Gibbons, R. Goldberg, R. Hornbeck, A. Luna, P. Murray-Rust, E. Neumann, O. Reubenacker, M. Samwald, M. van Iersel, S. Wimalaratne, K. Allen, B. Braun, M. Whirl-Carrillo, K. H. Cheung, Kam D. Dahlquist, A. Finney, M. Gillespie, E. Glass, L. Gong, M. Honig, O. Hubaut, D. Kane, S. Krupa, M. Kutmon, J. Leonard, D. Marks, D. Merberg, V. -

Specifications of Standards in Systems and Synthetic Biology
Specifications of Standards in Systems and Synthetic Biology: Status and Developments in 2019 Falk Schreiber1;2*, Gary D. Bader3, Padraig Gleeson4, Martin Golebiewski5, Michael Hucka6, Sarah M. Keating7, Matthias Konig¨ 8, Chris Myers9, David Nickerson10, Bjorn¨ Sommer1 and Dagmar Waltemath11 1Dep. of Computer and Information Science, University of Konstanz, Germany 2Faculty of IT, Monash University, Clayton, Australia 3The Donnelly Centre, University of Toronto, Canada 4Dep. of Neuroscience, Physiology and Pharmacology, University College London, UK 5Heidelberg Institute for Theoretical Studies (HITS), Germany 6California Institute of Technology, USA 7Heidelberg University, Germany 8Humboldt-University Berlin, Germany 9University of Utah, USA 10Auckland Bioengineering Institute, University of Auckland, New Zealand 11University Medicine Greifswald, Germany *To whom correspondence should be addressed. Email:[email protected] Abstract This special issue of the Journal of Integrative Bioinformatics presents an overview of COMBINE standards and their latest specifications. The standards cover representation formats for computational modeling in synthetic and systems biology and include BioPAX, CellML, NeuroML, SBML, SBGN, SBOL and SED-ML. The articles in this issue contain updated specifications of SBGN Process Description Level 1 Version 2, SBML Level 3 Core Version 2 Release 2, SBOL Version 2.3.0, and SBOL Visual Version 2.1. 1 Introduction Standards play an important role in Systems and Synthetic Biology. COMBINE (‘COmpu- tational Modeling in BIology’ NEtwork) [1, 2] is a formal entity that coordinates standards development in these fields of research, fosters and moderates discussions, designs and implements dissemination strategies, and organises two annual community meet- ings each year. HARMONY (Hackathons on Resources for Modeling in Biology) is a workshop and hackathon for the development of libraries, specifications and tool sup- port. -

Biomodels Database
BioModels Database Camille Laibe HARMONY 2012, 21-25th May 2012, Maastricht EBI is an Outstation of the European Molecular Biology Laboratory. What is BioModelsWho Database? are we? BioModels.net team Technology part of the Computational Systems Neurobiology group (Nicolas Le Novère) at EMBL-EBI Standards: Minimal Information Required In the Annotation of Models (MIRIAM), Minimal Information About a Simulation Experiment (MIASE), Systems Biology Graphical Notation (SBGN), ¼ Formats: Systems Biology Markup Language (SBML), Simulation Experiment Description Markup Language (SED-ML), ... Ontologies: Systems Biology Ontology (SBO), Kinetic Simulation Algorithm Ontology (KiSAO), TErminology for the Description of DYnamics (TEDDY), ¼ Services: BioModels Database, MIRIAM Registry, Identifiers.org, ... Tools: libSBML, JSBML, SBFC, SBMLeditor, ¼ 2 Basic (biochemical)What is BioModels model Database? example k k A+B 1 A_B 3 Ap+B k2 d[A]/dt = - k1[B][A] + k2[A_B] d[Ap]/dt = + k3[A_B] d[B]/dt = - k1[B][A] + k2[A_B] + k3[A_B] d[A_B]/dt = + k1[B][A] - k2[A_B] - k3[A_B] [x] 3 t How to build a model? biological model mathematical model simulation computational model4 Tyson et al (1991) PNAS 88(1):7328-32 What Whyis BioModels are models Database? useful? quantitative / dynamic understanding of biological systems integration of data from various scales make clear the current state of knowledge effective way of highlighting gaps in the knowledge prediction of the behaviour of systems under certain conditions sometimes the only tool available design novel