Towards Automating Structural Analysis of Complex Rna Molecules and Some Applications in Nanotechnology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Discovery of DNA Structure and Function: Watson and Crick By: Leslie A
01/08/2018 Discovery of DNA Double Helix: Watson and Crick | Learn Science at Scitable NUCLEIC ACID STRUCTURE AND FUNCTION | Lead Editor: Bob Moss Discovery of DNA Structure and Function: Watson and Crick By: Leslie A. Pray, Ph.D. © 2008 Nature Education Citation: Pray, L. (2008) Discovery of DNA structure and function: Watson and Crick. Nature Education 1(1):100 The landmark ideas of Watson and Crick relied heavily on the work of other scientists. What did the duo actually discover? Aa Aa Aa Many people believe that American biologist James Watson and English physicist Francis Crick discovered DNA in the 1950s. In reality, this is not the case. Rather, DNA was first identified in the late 1860s by Swiss chemist Friedrich Miescher. Then, in the decades following Miescher's discovery, other scientists--notably, Phoebus Levene and Erwin Chargaff--carried out a series of research efforts that revealed additional details about the DNA molecule, including its primary chemical components and the ways in which they joined with one another. Without the scientific foundation provided by these pioneers, Watson and Crick may never have reached their groundbreaking conclusion of 1953: that the DNA molecule exists in the form of a three-dimensional double helix. The First Piece of the Puzzle: Miescher Discovers DNA Although few people realize it, 1869 was a landmark year in genetic research, because it was the year in which Swiss physiological chemist Friedrich Miescher first identified what he called "nuclein" inside the nuclei of human white blood cells. (The term "nuclein" was later changed to "nucleic acid" and eventually to "deoxyribonucleic acid," or "DNA.") Miescher's plan was to isolate and characterize not the nuclein (which nobody at that time realized existed) but instead the protein components of leukocytes (white blood cells). -

Biochemistrystanford00kornrich.Pdf
University of California Berkeley Regional Oral History Office University of California The Bancroft Library Berkeley, California Program in the History of the Biosciences and Biotechnology Arthur Kornberg, M.D. BIOCHEMISTRY AT STANFORD, BIOTECHNOLOGY AT DNAX With an Introduction by Joshua Lederberg Interviews Conducted by Sally Smith Hughes, Ph.D. in 1997 Copyright 1998 by The Regents of the University of California Since 1954 the Regional Oral History Office has been interviewing leading participants in or well-placed witnesses to major events in the development of Northern California, the West, and the Nation. Oral history is a method of collecting historical information through tape-recorded interviews between a narrator with firsthand knowledge of historically significant events and a well- informed interviewer, with the goal of preserving substantive additions to the historical record. The tape recording is transcribed, lightly edited for continuity and clarity, and reviewed by the interviewee. The corrected manuscript is indexed, bound with photographs and illustrative materials, and placed in The Bancroft Library at the University of California, Berkeley, and in other research collections for scholarly use. Because it is primary material, oral history is not intended to present the final, verified, or complete narrative of events. It is a spoken account, offered by the interviewee in response to questioning, and as such it is reflective, partisan, deeply involved, and irreplaceable. ************************************ All uses of this manuscript are covered by a legal agreement between The Regents of the University of California and Arthur Kornberg, M.D., dated June 18, 1997. The manuscript is thereby made available for research purposes. All literary rights in the manuscript, including the right to publish, are reserved to The Bancroft Library of the University of California, Berkeley. -

Introduction and Historical Perspective
Chapter 1 Introduction and Historical Perspective “ Nothing in biology makes sense except in the light of evolution. ” modified by the developmental history of the organism, Theodosius Dobzhansky its physiology – from cellular to systems levels – and by the social and physical environment. Finally, behaviors are shaped through evolutionary forces of natural selection OVERVIEW that optimize survival and reproduction ( Figure 1.1 ). Truly, the study of behavior provides us with a window through Behavioral genetics aims to understand the genetic which we can view much of biology. mechanisms that enable the nervous system to direct Understanding behaviors requires a multidisciplinary appropriate interactions between organisms and their perspective, with regulation of gene expression at its core. social and physical environments. Early scientific The emerging field of behavioral genetics is still taking explorations of animal behavior defined the fields shape and its boundaries are still being defined. Behavioral of experimental psychology and classical ethology. genetics has evolved through the merger of experimental Behavioral genetics has emerged as an interdisciplin- psychology and classical ethology with evolutionary biol- ary science at the interface of experimental psychology, ogy and genetics, and also incorporates aspects of neuro- classical ethology, genetics, and neuroscience. This science ( Figure 1.2 ). To gain a perspective on the current chapter provides a brief overview of the emergence of definition of this field, it is helpful -
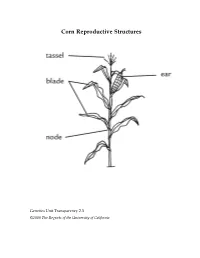
Corn Reproductive Structures
Corn Reproductive Structures Genetics Unit Transparency 2.3 ©2008 The Regents of the University of California Creating a Punnett Square Genetics Unit Transparency 2.4 ©2008 The Regents of the University of California Case Study Summary Sheet Case Study Type of Benefits Risks and concerns Remaining questions Genetic Modification Genetics Unit Transparency 4.1 ©2008 The Regents of the University of California Class Data for Rice Breeding Simulation aromatic, non-aromatic, aromatic, non-aromatic ,flood-tolear nt flood-tolerant flood-intolerant flood-intolerant AAFF AAFf AaFF AaFf aaFF aaFf AAff Aaff aaff Group 1 2 3 4 5 6 7 8 Genetics Unit Transparency 5.1 ©2008 The Regents of the University of California Three Types of Cells Genetics Unit Transparency 6.1 ©2008 The Regents of the University of California DNA base percentages in a variety of organisms Source of A T G C DNA Human 31.0% 31.5% 19.1% 18.4% Mouse 29.1% 29.0% 21.1% 21.1% Frog 26.3% 26.4% 23.5% 23.8% Fruit fly 27.3% 27.6% 22.5% 22.5% Corn 25.6% 25.3% 24.5% 24.6% E. coli 24.6% 24.3% 25.5% 25.6% Genetics Unit Transparency 7.1 ©2008 The Regents of the University of California Matthew Meselson and Franklin Stahl’s Experiment to Investigate DNA Replication First: Scientists James Watson and Francis Crick propose a method of semi-conservative replication in their paper on the structure of DNA. However, they have no data to support their hypothesis. Next: Matthew Meselson and Franklin Stahl use the procedure that follows to investigate DNA created by the process of DNA Replication. -

1 History for Canonical and Non-Canonical Structures of Nucleic Acids
1 1 History for Canonical and Non-canonical Structures of Nucleic Acids The main points of the learning: Understand canonical and non-canonical structures of nucleic acids and think of historical scientists in the research field of nucleic acids. 1.1 Introduction This book is to interpret the non-canonical structures and their stabilities of nucleic acids from the viewpoint of the chemistry and study their biological significances. There is more than 60 years’ history after the discovery of the double helix DNA structure by James Dewey Watson and Francis Harry Compton Crick in 1953, and chemical biology of nucleic acids is facing a new aspect today. Through this book, I expect that readers understand how the uncommon structure of nucleic acids became one of the common structures that fascinate us now. In this chapter, I introduce the history of nucleic acid structures and the perspective of research for non-canonical nucleic acid structures (see also Chapter 15). 1.2 History of Duplex The opening of the history of genetics was mainly done by three researchers. Charles Robert Darwin, who was a scientist of natural science, pioneered genetics. The proposition of genetic concept is indicated in his book On the Origin of Species published in 1859. He indicated the theory of biological evolution, which is the basic scientific hypothesis of natural diversity. In other words, he proposed biological evolution, which changed among individuals by adapting to the environment and be passed on to the next generation. However, that was still a primitive idea for the genetic concept. After that, Gregor Johann Mendel, who was a priest in Brno, Czech Republic, confirmed the mechanism of gene evolution by using “factor” inherited from parent to children using pea plant in 1865. -

DNA: the Timeline and Evidence of Discovery
1/19/2017 DNA: The Timeline and Evidence of Discovery Interactive Click and Learn (Ann Brokaw Rocky River High School) Introduction For almost a century, many scientists paved the way to the ultimate discovery of DNA and its double helix structure. Without the work of these pioneering scientists, Watson and Crick may never have made their ground-breaking double helix model, published in 1953. The knowledge of how genetic material is stored and copied in this molecule gave rise to a new way of looking at and manipulating biological processes, called molecular biology. The breakthrough changed the face of biology and our lives forever. Watch The Double Helix short film (approximately 15 minutes) – hyperlinked here. 1 1/19/2017 1865 The Garden Pea 1865 The Garden Pea In 1865, Gregor Mendel established the foundation of genetics by unraveling the basic principles of heredity, though his work would not be recognized as “revolutionary” until after his death. By studying the common garden pea plant, Mendel demonstrated the inheritance of “discrete units” and introduced the idea that the inheritance of these units from generation to generation follows particular patterns. These patterns are now referred to as the “Laws of Mendelian Inheritance.” 2 1/19/2017 1869 The Isolation of “Nuclein” 1869 Isolated Nuclein Friedrich Miescher, a Swiss researcher, noticed an unknown precipitate in his work with white blood cells. Upon isolating the material, he noted that it resisted protein-digesting enzymes. Why is it important that the material was not digested by the enzymes? Further work led him to the discovery that the substance contained carbon, hydrogen, nitrogen and large amounts of phosphorus with no sulfur. -

James A. Mccloskey, Jr
CHEMICAL HERITAGE FOUNDATION JAMES A. MCCLOSKEY, JR. Transcript of Interviews Conducted by Michael A. Grayson at the McCloskeys’ Home Helotes, Texas on 19 and 20 March 2012 (With Subsequent Corrections and Additions) James A. McCloskey, Jr. ACKNOWLEDGMENT This oral history is one in a series initiated by the Chemical Heritage Foundation on behalf of the American Society for Mass Spectrometry. The series documents the personal perspectives of individuals related to the advancement of mass spectrometric instrumentation, and records the human dimensions of the growth of mass spectrometry in academic, industrial, and governmental laboratories during the twentieth century. This project is made possible through the generous support of the American Society for Mass Spectrometry. This oral history is designated Free Access. Please note: Users citing this interview for purposes of publication are obliged under the terms of the Chemical Heritage Foundation (CHF) Center for Oral History to credit CHF using the format below: James A. McCloskey, Jr., interview by Michael A. Grayson at the McCloskeys’ home, Helotes, Texas, 19-20 March 2012 (Philadelphia: Chemical Heritage Foundation, Oral History Transcript # 0702). Chemical Heritage Foundation Center for Oral History 315 Chestnut Street Philadelphia, Pennsylvania 19106 The Chemical Heritage Foundation (CHF) serves the community of the chemical and molecular sciences, and the wider public, by treasuring the past, educating the present, and inspiring the future. CHF maintains a world-class collection of materials that document the history and heritage of the chemical and molecular sciences, technologies, and industries; encourages research in CHF collections; and carries out a program of outreach and interpretation in order to advance an understanding of the role of the chemical and molecular sciences, technologies, and industries in shaping society. -

Structure of DNA &
Structure Of DNA & RNA By Himanshu Dev VMMC & SJH DNA DNA Deoxyribonucleic acid DNA - a polymer of deoxyribo- nucleotides. Usually double stranded. And have double-helix structure. found in chromosomes, mitochondria and chloroplasts. It acts as the genetic material in most of the organisms. Carries the genetic information A Few Key Events Led to the Discovery of the Structure of DNA DNA as an acidic substance present in nucleus was first identified by Friedrich Meischer in 1868. He named it as ‘Nuclein’. Friedrich Meischer In 1953 , James Watson and Francis Crick, described a very simple but famous Double Helix model for the structure of DNA. FRANCIS CRICK AND JAMES WATSON The scientific framework for their breakthrough was provided by other scientists including Linus Pauling Rosalind Franklin and Maurice Wilkins Erwin Chargaff Rosalind Franklin She worked in same laboratory as Maurice Wilkins. She study X-ray diffraction to study wet fibers of DNA. X-ray diffraction of wet DNA fibers The diffraction pattern is interpreted X Ray (using mathematical theory) Crystallography This can ultimately provide Rosalind information concerning the structure Franklin’s photo of the molecule She made marked advances in X-ray diffraction techniques with DNA The diffraction pattern she obtained suggested several structural features of DNA Helical More than one strand 10 base pairs per complete turn Rosalind Franklin Maurice Wilkins DNA Structure DNA structure is often divided into four different levels primary, secondary, tertiary -

Jewels in the Crown
Jewels in the crown CSHL’s 8 Nobel laureates Eight scientists who have worked at Cold Max Delbrück and Salvador Luria Spring Harbor Laboratory over its first 125 years have earned the ultimate Beginning in 1941, two scientists, both refugees of European honor, the Nobel Prize for Physiology fascism, began spending their summers doing research at Cold or Medicine. Some have been full- Spring Harbor. In this idyllic setting, the pair—who had full-time time faculty members; others came appointments elsewhere—explored the deep mystery of genetics to the Lab to do summer research by exploiting the simplicity of tiny viruses called bacteriophages, or a postdoctoral fellowship. Two, or phages, which infect bacteria. Max Delbrück and Salvador who performed experiments at Luria, original protagonists in what came to be called the Phage the Lab as part of the historic Group, were at the center of a movement whose members made Phage Group, later served as seminal discoveries that launched the revolutionary field of mo- Directors. lecular genetics. Their distinctive math- and physics-oriented ap- Peter Tarr proach to biology, partly a reflection of Delbrück’s physics train- ing, was propagated far and wide via the famous Phage Course that Delbrück first taught in 1945. The famous Luria-Delbrück experiment of 1943 showed that genetic mutations occur ran- domly in bacteria, not necessarily in response to selection. The pair also showed that resistance was a heritable trait in the tiny organisms. Delbrück and Luria, along with Alfred Hershey, were awarded a Nobel Prize in 1969 “for their discoveries concerning the replication mechanism and the genetic structure of viruses.” Barbara McClintock Alfred Hershey Today we know that “jumping genes”—transposable elements (TEs)—are littered everywhere, like so much Alfred Hershey first came to Cold Spring Harbor to participate in Phage Group wreckage, in the chromosomes of every organism. -

The Great-Grandmother of LUCA (Last Universal Common Ancestor)
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 4 June 2018 doi:10.20944/preprints201806.0035.v1 Be introduced to the First Universal Common Ancestor (FUCA): the great-grandmother of LUCA (Last Universal Common Ancestor) Francisco Prosdocimi1*, Marco V José2 and Sávio Torres de Farias3* 1 Laboratório de Biologia Teórica e de Sistemas, Instituto de Bioquímica Médica Leopoldo de Meis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brasil. 2 Theoretical Biology Group, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México, Ciudad Universitaria, 04510 CDMX, Mexico. 3 Laboratório de Genética Evolutiva Paulo Leminsk, Departamento de Biologia Molecular, Universidade Federal da Paraíba, João Pessoa, Paraíba, Brasil. * Correspondence: [email protected]; [email protected] Abstract The existence of a common ancestor to all living organisms in Earth is a necessary corollary of Darwin idea of common ancestry. The Last Universal Common Ancestor (LUCA) has been normally considered as the ancestor of cellular organisms that originated the three domains of life: Bacteria, Archaea and Eukarya. Recent studies about the nature of LUCA indicate that this first organism should present hundreds of genes and a complex metabolism. Trying to bring another of Darwin ideas into the origins of life discussion, we went back into the prebiotic chemistry trying to understand how LUCA could be originated 1 © 2018 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 4 June 2018 doi:10.20944/preprints201806.0035.v1 under gradualist assumptions. Along this line of reasoning, it became clear to us that the definition of another ancestral should be of particular relevance to the understanding about the emergence of biological systems. -

Genes, Genomes and Genetic Analysis
© Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION UNIT 1 © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FORDefining SALE OR DISTRIBUTION and WorkingNOT FOR SALE OR DISTRIBUTION with Genes © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION Chapter 1 Genes, Genomes, and Genetic Analysis Chapter 2 DNA Structure and Genetic Variation © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Molekuul/Science Photo Library/Getty Images. © Jones & Bartlett Learning, LLC © Jones & Bartlett Learning, LLC NOT FOR SALE OR DISTRIBUTION NOT FOR SALE OR DISTRIBUTION © Jones & Bartlett Learning, LLC. NOT FOR SALE OR DISTRIBUTION 9781284136609_CH01_Hartl.indd 1 08/11/17 8:50 am © Jones & Bartlett Learning, LLC -

Beyond the Big Bang • the Amazon's Lost Civilizations • the Truth
SFI Bulletin winter 2006, vol. 21 #1 Beyond the Big Bang • The Amazon’s Lost Civilizations • The Truth Behind Lying The Bulletin of the Santa Fe Institute is published by SFI to keep its friends and supporters informed about its work. The Santa Fe Institute is a private, independent, multidiscipli- nary research and education center founded in 1984. Since its founding, SFI has devoted itself to creating a new kind of sci- entific research community, pursuing emerging synthesis in science. Operating as a visiting institution, SFI seeks to cat- alyze new collaborative, multidisciplinary research; to break down the barriers between the traditional disciplines; to spread its ideas and methodologies to other institutions; and to encourage the practical application of its results. Published by the Santa Fe Institute 1399 Hyde Park Road Santa Fe, New Mexico 87501, USA phone (505) 984-8800 fax (505) 982-0565 home page: http://www.santafe.edu Note: The SFI Bulletin may be read at the website: www.santafe.edu/sfi/publications/Bulletin/. If you would prefer to read the Bulletin on your computer rather than receive a printed version, contact Patrisia Brunello at 505/984-8800, Ext. 2700 or [email protected]. EDITORIAL STAFF: Ginger Richardson Lesley S. King Andi Sutherland CONTRIBUTORS: Brooke Harrington Janet Yagoda Shagam Julian Smith Janet Stites James Trefil DESIGN & PRODUCTION: Paula Eastwood PHOTO: ROBERT BUELTEMAN ©2004 BUELTEMAN PHOTO: ROBERT SFI Bulletin Winter 2006 TOCtable of contents 3 A Deceptively Simple Formula 2 How Life Began 3 From