Λ Recombineering Used to Engineer the Genome of Phage T7
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Concatemerization of Synthetic Oligonucleotides Assembly and Ligand Interactions
THESIS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY CONCATEMERIZATION OF SYNTHETIC OLIGONUCLEOTIDES ASSEMBLY AND LIGAND INTERACTIONS LU SUN Department of Chemical and Biological Engineering CHALMERS UNIVERSITY OF TECHNOLOGY GÖTEBORG, SWEDEN 2014 Concatemerization of Synthetic Oligonucleotides Assembly and Ligand Interactions LU SUN ISBN 978-91-7597-092-9 © LU SUN, 2014 Doktorsavhandlingar vid Chalmers tekniska högskola Ny serie nr 3773 ISSN 0346-718X Department of Chemical and Biological Engineering Chalmers University of Technology SE-412 96 Göteborg Sweden Telephone + 46 (0)31-772 1000 Cover image: Two ways of forming DNA concatemers. (Top right) Self-assembly of oligonucleotides in free solution. (Down) A three-step sequential assembly of DNA concatemer layer on a planar surface and RAD51 induced DNA layer extension. Back cover photo: © Keling Bi Printed by Chalmers Reproservice Göteborg, Sweden 2014 ii Concatemerization of Synthetic Oligonucleotides Assembly and Ligand Interactions LU SUN Department of Chemical and Biological Engineering Chalmers University of Technology ABSTRACT DNA nanotechnology has become an important research field because of its advantages in high predictability and accuracy of base paring recognition. For example, the DNA concatemer, one of the simplest DNA constructs in shape, has been used to enhance signals in biosensing. In this thesis, concatemers were designed and characterized towards sequence-specific target amplifiers for single-molecule mechanical studies. The project firstly focuses on exploring how to obtain concatemers of satisfactory length by self- assembly in bulk. Concatemers formed in solution by mixing of the different components are characterized using gel electrophoresis and AFM. Experimental results demonstrate that the concatemerization yield could be increased primarily by increasing the ionic strength, and linear concatemers of expected length could be separated from mixed sizes and shapes. -

The Rate of T7 Phage-Mediated Lysis of Escherichia Coli Growing in Exponential Phase Is Not Affected by Deletion of Rpos
Undergraduate Journal of Experimental Microbiology and Immunology (UJEMI) Vol. 25:1- 9 Copyright © September 2020, M&I UBC The rate of T7 phage-mediated lysis of Escherichia coli growing in exponential phase is not affected by deletion of rpoS Jingyao Zhu, Cai Lan Jennifer Huang, Kamand Doraki, Bryan Lee Department of Microbiology and Immunology, University of British Columbia, Vancouver, British Columbia, Canada SUMMARY RpoS is an Escherichia coli sigma factor shown to regulate genes in response to various environmental and internal stressors. Previous studies have investigated the role of RpoS in a mechanism known as a cross-protection, in which prior exposure to one stressor leads to increased tolerance to a subsequent stressor. Pre-treatment with subinhibitory levels of antibiotics have been used in several studies in an attempt to elicit a delay in phage-lysis, with varying and contradicting results. We propose that the delayed T7-mediated lysis phenotype may be growth phase dependent as the stress response sigma factor RpoS is known to be expressed in stationary phase. Here, we attempted to perform phage lysis assays on wild-type (WT) and rpoS knockout strains of Escherichia coli strains growing in stationary and exponential phase, to look for differences in the time to observe bacteriophage-mediated lysis. We did not observe differences in the time of phage-mediated lysis between wild type and rpoS knock-out strains of E. coli growing in exponential phase. An attempt was made to compare phage-mediated lysis time of E. coli grown in exponential and stationary phase, however, technical issues related to normalizing optical density prevented meaningful comparisons. -

CRISPR/Cas9 Mediated T7 RNA Polymerase Gene Knock-In in E. Coli BW25113 Makes T7 Expression System Work Efciently
CRISPR/Cas9 mediated T7 RNA polymerase gene knock-in in E. coli BW25113 makes T7 expression system work eciently Changchuan Ye China Agricultural University https://orcid.org/0000-0002-0683-5290 Xi Chen China Agricultural University Mengjie Yang National Feed Engineering Technology Research Centre Xiangfang Zeng China Agricultural University College of Animal Science and Technology Shiyan Qiao ( [email protected] ) China Agricultural University College of Animal Science and Technology https://orcid.org/0000-0003-4434-318X Research Keywords: E. coli, CRISPR/cas9, T7 Expression System, Fluorescent Protein, 5-Aminolevulinic Acid, promoter variants Posted Date: March 24th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-339217/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Journal of Biological Engineering on August 12th, 2021. See the published version at https://doi.org/10.1186/s13036-021-00270-9. Page 1/23 Abstract T7 Expression System is a common method of ensuring tight control and high-level induced expression. However, this system can only work in some bacterial strains in which the T7 RNA Polymerase gene resides in the chromosome. In this study, we successfully introduced a chromosomal copy of the T7 RNA Polymerase gene under control of the lacUV5 promoter into Escherichia coli BW25113. The T7 Expression System worked eciently in this mutant strain named BW25113-T7. We demonstrated that this mutant strain could satisfactorily produce 5-Aminolevulinic Acid via C5 pathway. A nal study was designed to enhance the controllability of T7 Expression System in this mutant strain by constructing a T7 Promoter Variants Library. -

Co-Inoculation of Escherichia Coli B23 by T4 and T7 Bacteriophages Results in Competition Shown by an Overall Drop in Phage Progeny
Journal of Experimental Microbiology and Immunology (JEMI) Vol. 18: 156 – 161 Copyright © April 2014, M&I UBC Co-inoculation of Escherichia coli B23 by T4 and T7 bacteriophages results in competition shown by an overall drop in phage progeny Khue-Tu Nguyen, Karen Simmons, Igor Tatarnikov Department Microbiology & Immunology, University of British Columbia Co-inoculation and co-infection are phenomena that occur naturally in the environment that have implications on. In order to explore competition between T4 and T7 phages, one-step assays were carried out on T4 and T7 mono- infected samples, and a sample co-inoculated by both T4 and T7. Mono-infections were performed at an MOI of 5 in order to ensure infection, and the co-inoculation had an MOI of 5 for both T4 and T7 resulting in a total MOI of 10. It was found that at 90 min post co-inoculation, the overall titer of both phages decreased with respect to their mono-infected controls, 97% for T4 and 98% for T7. This suggested that actual co-infection could have occurred and competition was carried out within the host cell. This effect demonstrated that even though there seems to be interference between the two phages in co-inoculation, neither phage out-competes the other. T4 and T7 are well-studied bacteriophage capable of from the Microbiology 421 culture collection from the infecting Escherichia coli (E. coli) (1, 2). They contain department of Microbiology and Immunology, University of double-stranded, linear, DNA genomes, with T4 having British Columbia. approximately 169 kbp coding for around 300 genes (1,3) PCR. -

Genomic Analysis of Bacteriophages SP6 and K1-5, an Estranged Subgroup of the T7 Supergroup
doi:10.1016/j.jmb.2003.11.035 J. Mol. Biol. (2004) 335, 1151–1171 Genomic Analysis of Bacteriophages SP6 and K1-5, an Estranged Subgroup of the T7 Supergroup D. Scholl1*, J. Kieleczawa2, P. Kemp3, J. Rush4, C. C. Richardson4 C. Merril1, S. Adhya5 and I. J. Molineux3,6 1Section of Biochemical We have determined the genome sequences of two closely related lytic Genetics, The National bacteriophages, SP6 and K1-5, which infect Salmonella typhimurium LT2 Institute of Mental Health and Escherichia coli serotypes K1 and K5, respectively. The genome organ- NIH, 9000 Rockville Pike ization of these phages is almost identical with the notable exception of Bethesda, MD 20895, USA the tail fiber genes that confer the different host specificities. The two phages have diverged extensively at the nucleotide level but they are still 2Wyeth/Genetics Institute more closely related to each other than either is to any other phage cur- Cambridge, MA 02140, USA rently characterized. The SP6 and K1-5 genomes contain, respectively, 3Microbiology and Molecular 43,769 bp and 44,385 bp, with 174 bp and 234 bp direct terminal repeats. Genetics, University of Texas About half of the 105 putative open reading frames in the two genomes Austin, TX 78712, USA combined show no significant similarity to database proteins with a 4 known or predicted function that is obviously beneficial for growth of a Department of Biological bacteriophage. The overall genome organization of SP6 and K1-5 is com- Chemistry and Molecular parable to that of the T7 group of phages, although the specific order of Pharmacology, Harvard genes coding for DNA metabolism functions has not been conserved. -

A Highly Efficient Recombineering-Based Method for Generating Conditional Knockout Mutations Pentao Liu, Nancy A
Methods A Highly Efficient Recombineering-Based Method for Generating Conditional Knockout Mutations Pentao Liu, Nancy A. Jenkins, and Neal G. Copeland1 Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702, USA Phage-based Escherichia coli homologous recombination systems have recently been developed that now make it possible to subclone or modify DNA cloned into plasmids, BACs, or PACs without the need for restriction enzymes or DNA ligases. This new form of chromosome engineering, termed recombineering, has many different uses for functional genomic studies. Here we describe a new recombineering-based method for generating conditional mouse knockout (cko) mutations. This method uses homologous recombination mediated by the phage Red proteins, to subclone DNA from BACs into high-copy plasmids by gaprepair,and together with Cre or Flpe recombinases, to introduce loxPorFRT sites into the subcloned DNA. Unlike other methods that use short 45–55-bpregions of homology for recombineering, our metho d uses much longer regions of homology. We also make use of several new E. coli strains, in which the proteins required for recombination are expressed from a defective temperature-sensitive prophage, and the Cre or Flpe recombinases from an arabinose-inducible promoter. We also describe two new Neo selection cassettes that work well in both E. coli and mouse ES cells. Our method is fast, efficient, and reliable and makes it possible to generate cko-targeting vectors in less than 2 wk. This method should also facilitate the generation of knock-in mutations and transgene constructs, as well as expedite the analysis of regulatory elements and functional domains in or near genes. -

Ancestral Gene Acquisition As the Key to Virulence Potential in Environmental Vibrio Populations
The ISME Journal (2018) 12:2954–2966 https://doi.org/10.1038/s41396-018-0245-3 ARTICLE Ancestral gene acquisition as the key to virulence potential in environmental Vibrio populations 1,2 1,2 1,2 1,2 2 1,3 Maxime Bruto ● Yannick Labreuche ● Adèle James ● Damien Piel ● Sabine Chenivesse ● Bruno Petton ● 4 1,2 Martin F. Polz ● Frédérique Le Roux Received: 4 March 2018 / Revised: 30 June 2018 / Accepted: 6 July 2018 / Published online: 2 August 2018 © The Author(s) 2018. This article is published with open access Abstract Diseases of marine animals caused by bacteria of the genus Vibrio are on the rise worldwide. Understanding the eco- evolutionary dynamics of these infectious agents is important for predicting and managing these diseases. Yet, compared to Vibrio infecting humans, knowledge of their role as animal pathogens is scarce. Here we ask how widespread is virulence among ecologically differentiated Vibrio populations, and what is the nature and frequency of virulence genes within these populations? We use a combination of population genomics and molecular genetics to assay hundreds of Vibrio strains for their virulence in the oyster Crassostrea gigas, a unique animal model that allows high-throughput infection assays. We 1234567890();,: 1234567890();,: show that within the diverse Splendidus clade, virulence represents an ancestral trait but has been lost from several populations. Two loci are necessary for virulence, the first being widely distributed across the Splendidus clade and consisting of an exported conserved protein (R5.7). The second is a MARTX toxin cluster, which only occurs within V. splendidus and is for the first time associated with virulence in marine invertebrates. -
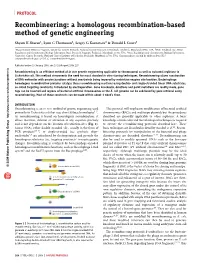
A Homologous Recombination-Based Method of Genetic Engineering
PROTOCOL Recombineering: a homologous recombination-based method of genetic engineering Shyam K Sharan1, Lynn C Thomason2, Sergey G Kuznetsov1 & Donald L Court3 1Mouse Cancer Genetics Program, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702, USA. 2SAIC-Frederick Inc., Gene Regulation and Chromosome Biology Laboratory, Basic Research Program, Frederick, Maryland, 21702, USA. 3Gene Regulation and Chromosome Biology Laboratory, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702, USA. Correspondence should be addressed to S.K.S. ([email protected]) or D.L.C. ([email protected]). Published online 29 January 2009; doi:10.1038/nprot.2008.227 Recombineering is an efficient method of in vivo genetic engineering applicable to chromosomal as well as episomal replicons in Escherichia coli. This method circumvents the need for most standard in vitro cloning techniques. Recombineering allows construction of DNA molecules with precise junctions without constraints being imposed by restriction enzyme site location. Bacteriophage homologous recombination proteins catalyze these recombineering reactions using double- and single-stranded linear DNA substrates, natureprotocols so-called targeting constructs, introduced by electroporation. Gene knockouts, deletions and point mutations are readily made, gene / m tags can be inserted and regions of bacterial artificial chromosomes or the E. coli genome can be subcloned by gene retrieval using o c . recombineering. Most of these constructs can be made within about 1 week’s time. e r u t a n . INTRODUCTION w w Recombineering is an in vivo method of genetic engineering used This protocol will emphasize modification of bacterial artificial w / / 1–5 : primarily in Escherichia coli that uses short 50 base homologies . -

Bacterial Artificial Chromosome Mutagenesis Using Recombineering
Hindawi Publishing Corporation Journal of Biomedicine and Biotechnology Volume 2011, Article ID 971296, 10 pages doi:10.1155/2011/971296 Review Article Bacterial Artificial Chromosome Mutagenesis Using Recombineering Kumaran Narayanan1, 2 and Qingwen Chen2 1 Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY 10029, USA 2 School of Science, Monash University, Sunway Campus, Room 2-5-29, Bandar Sunway, 46150, Malaysia Correspondence should be addressed to Kumaran Narayanan, [email protected] Received 2 August 2010; Accepted 21 October 2010 Academic Editor: Masamitsu Yamaguchi Copyright © 2011 K. Narayanan and Q. Chen. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Gene expression from bacterial artificial chromosome (BAC) clones has been demonstrated to facilitate physiologically relevant levels compared to viral and nonviral cDNA vectors. BACs are large enough to transfer intact genes in their native chromosomal setting together with flanking regulatory elements to provide all the signals for correct spatiotemporal gene expression. Until recently, the use of BACs for functional studies has been limited because their large size has inherently presented a major obstacle for introducing modifications using conventional genetic engineering strategies. The development of in vivo homologous recombination strategies based on recombineering in E. coli has helped resolve this problem by enabling facile engineering of high molecular weight BAC DNA without dependence on suitably placed restriction enzymes or cloning steps. These techniques have considerably expanded the possibilities for studying functional genetics using BACs in vitro and in vivo. -

Genetic Engineering Using Homologous Recombination1
17 Oct 2002 13:49 AR AR174-GE36-14.tex AR174-GE36-14.SGM LaTeX2e(2002/01/18) P1: IBC 10.1146/annurev.genet.36.061102.093104 Annu. Rev. Genet. 2002. 36:361–88 doi: 10.1146/annurev.genet.36.061102.093104 GENETIC ENGINEERING USING HOMOLOGOUS RECOMBINATION1 Donald L. Court, James A. Sawitzke, and Lynn C. Thomason Gene Regulation and Chromosome Biology Laboratory, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702; e-mail: [email protected], [email protected], [email protected] Key Words DNA replication forks, strand annealing, in vivo cloning, oligo recombination, recombineering ■ Abstract In the past few years, in vivo technologies have emerged that, due to their efficiency and simplicity, may one day replace standard genetic engineering tech- niques. Constructs can be made on plasmids or directly on the Escherichia coli chromo- some from PCR products or synthetic oligonucleotides by homologous recombination. This is possible because bacteriophage-encoded recombination functions efficiently re- combine sequences with homologies as short as 35 to 50 base pairs. This technology, termed recombineering, is providing new ways to modify genes and segments of the chromosome. This review describes not only recombineering and its applications, but also summarizes homologous recombination in E. coli and early uses of homologous recombination to modify the bacterial chromosome. Finally, based on the premise that phage-mediated recombination functions act at replication forks, specific molecular models are -

Bacteriophage T7 Gene 2.5 Protein: an Essential Protein for DNA Replication (DNA Binding Protein/Recombination/T7 DNA Polymerase) YOUNG TAE KIM* and CHARLES C
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 10173-10177, November 1993 Biochemistry Bacteriophage T7 gene 2.5 protein: An essential protein for DNA replication (DNA binding protein/recombination/T7 DNA polymerase) YOUNG TAE KIM* AND CHARLES C. RICHARDSON Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115 Contributed by Charles C. Richardson, July 22, 1993 ABSTRACT The product of gene 2.5 of bacteriophage T7, 15). Both direct and indirect evidence support an interaction a single-stranded DNA binding protein, physically interacts with between these essential replication proteins and the T7 gene the phage-encoded gene S protein (DNA polymerase) and gene 2.5 protein. 4 proteins (helicase and primase) and stimulates their activities. T7 gene 2.5 protein stimulates DNA synthesis catalyzed by Genetic analysis ofT7 phage defective in gene 2.5 shows that the the T7 DNA polymerase/thioredoxin complex on single- gene 2.5 protein is essential for T7 DNA replication and growth. stranded DNA templates (4, 5, 11, 14) and increases the T7 phages thatcontain null mutants ofgene2.5 were constructed processivity ofthe reaction (11). It has been shown by affinity by homologous recombination. These gene 2.5 null mutants chromatography and fluorescence emission anisotropy that contain either a deletion ofgene2.5 (T7A2.5) or an insertion into T7 DNA polymerase and gene 2.5 protein physically interact gene 2.5 and cannot grow in Escherichia coli (efficiency of with a dissociation constant of 1.1 ,uM (11). Similarly, inter- plating, <10-8). After infection of E. coli with T7A2.5, host action of the T7 helicase/primase with gene 2.5 protein has DNA synthesis is shut off, and phage DNA synthesis is reduced been inferred from the ability ofgene 2.5 protein to stimulate to <1% ofphage DNA synthesis in wild-type T7-infected E. -

Improved Bacterial Recombineering by Parallelized Protein Discovery
Improved bacterial recombineering by parallelized protein discovery Timothy M. Wanniera,1, Akos Nyergesa,b, Helene M. Kuchwaraa, Márton Czikkelyb, Dávid Baloghb, Gabriel T. Filsingera, Nathaniel C. Bordersa, Christopher J. Gregga, Marc J. Lajoiea, Xavier Riosa, Csaba Pálb, and George M. Churcha,1 aDepartment of Genetics, Harvard Medical School, Boston, MA 02115; and bSynthetic and Systems Biology Unit, Institute of Biochemistry, Biological Research Centre, Szeged HU-6726, Hungary Edited by Susan Gottesman, National Institutes of Health, Bethesda, MD, and approved April 17, 2020 (received for review January 30, 2020) Exploiting bacteriophage-derived homologous recombination pro- recombineering, is the molecular mechanism that drives MAGE cesses has enabled precise, multiplex editing of microbial genomes and DIvERGE (4, 32, 33). This method was first described in and the construction of billions of customized genetic variants in a E. coli, and is most commonly promoted by the expression of bet single day. The techniques that enable this, multiplex automated ge- (here referred to as Redβ) from the Red operon of Escherichia nome engineering (MAGE) and directed evolution with random ge- phage λ (5, 6, 34). Redβ is an ssDNA-annealing protein (SSAP) nomic mutations (DIvERGE), are however, currently limited to a whose role in recombineering is to anneal ssDNA to compli- handful of microorganisms for which single-stranded DNA-annealing mentary genomic DNA at the replication fork. Although im- proteins (SSAPs) that promote efficient recombineering have been provements to recombineering efficiency have been made (7–9), identified. Thus, to enable genome-scale engineering in new hosts, the core protein machinery has remained constant, with Redβ efficient SSAPs must first be found.