Hereditary Hemolytic Anemia Cascade
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phenotypic and Molecular Genetic Analysis of Pyruvate Kinase Deficiency in a Tunisian Family
The Egyptian Journal of Medical Human Genetics (2016) 17, 265–270 HOSTED BY Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com ORIGINAL ARTICLE Phenotypic and molecular genetic analysis of Pyruvate Kinase deficiency in a Tunisian family Jaouani Mouna a,1,*, Hamdi Nadia a,1, Chaouch Leila a, Kalai Miniar a, Mellouli Fethi b, Darragi Imen a, Boudriga Imen a, Chaouachi Dorra a, Bejaoui Mohamed b, Abbes Salem a a Laboratory of Molecular and Cellular Hematology, Pasteur Institute, Tunis, Tunisia b Service d’Immuno-He´matologie Pe´diatrique, Centre National de Greffe de Moelle Osseuse, Tunis, Tunisia Received 9 July 2015; accepted 6 September 2015 Available online 26 September 2015 KEYWORDS Abstract Pyruvate Kinase (PK) deficiency is the most frequent red cell enzymatic defect responsi- Pyruvate Kinase deficiency; ble for hereditary non-spherocytic hemolytic anemia. The disease has been studied in several ethnic Phenotypic and molecular groups. However, it is yet an unknown pathology in Tunisia. We report here, the phenotypic and investigation; molecular investigation of PK deficiency in a Tunisian family. Hemolytic anemia; This study was carried out on two Tunisian brothers and members of their family. Hematolog- Hydrops fetalis; ical, biochemical analysis and erythrocyte PK activity were performed. The molecular characteriza- PKLR mutation tion was carried out by gene sequencing technique. The first patient died few hours after birth by hydrops fetalis, the second one presented with neonatal jaundice and severe anemia necessitating urgent blood transfusion. This severe clinical pic- ture is the result of a homozygous mutation of PKLR gene at exon 8 (c.1079G>A; p.Cys360Tyr). -

Non-Commercial Use Only
only use Non-commercial 14th International Conference on Thalassaemia and Other Haemoglobinopathies 16th TIF Conference for Patients and Parents 17-19 November 2017 • Grand Hotel Palace, Thessaloniki, Greece only use For thalassemia patients with chronic transfusional iron overload... Make a lasting impression with EXJADENon-commercial film-coated tablets The efficacy of deferasirox in a convenient once-daily film-coated tablet Please see your local Novartis representative for Full Product Information Reference: EXJADE® film-coated tablets [EU Summary of Product Characteristics]. Novartis; August 2017. Important note: Before prescribing, consult full prescribing information. iron after having achieved a satisfactory body iron level and therefore retreatment cannot be recommended. ♦ Maximum daily dose is 14 mg/kg body weight. ♦ In pediatric patients the Presentation: Dispersible tablets containing 125 mg, 250 mg or 500 mg of deferasirox. dosing should not exceed 7 mg/kg; closer monitoring of LIC and serum ferritin is essential Film-coated tablets containing 90 mg, 180 mg or 360 mg of deferasirox. to avoid overchelation; in addition to monthly serum ferritin assessments, LIC should be Indications: For the treatment of chronic iron overload due to frequent blood transfusions monitored every 3 months when serum ferritin is ≤800 micrograms/l. (≥7 ml/kg/month of packed red blood cells) in patients with beta-thalassemia major aged Dosage: Special population ♦ In moderate hepatic impairment (Child-Pugh B) dose should 6 years and older. ♦ Also indicated for the treatment of chronic iron overload due to blood not exceed 50% of the normal dose. Should not be used in severe hepatic impairment transfusions when deferoxamine therapy is contraindicated or inadequate in the following (Child-Pugh C). -

Predictors of Autoimmune Hemolytic Anemia in Beta-Thalassemia
Blood Cells, Molecules and Diseases 79 (2019) 102342 Contents lists available at ScienceDirect Blood Cells, Molecules and Diseases journal homepage: www.elsevier.com/locate/bcmd Predictors of autoimmune hemolytic anemia in beta-thalassemia patients with underlying red blood cells autoantibodies T ⁎ Monia Ben Khaleda,b, , Monia Ouedernia,b, Nessrine Sahlia,b, Nawel Dhouibb, Ahmed Ben Abdelazizc, Samia Rekayaa,b, Ridha Koukia,b, Houda Kaabid, Hmida Slamad, Fethi Melloulia,b, Mohamed Bejaouia,b a Faculty of Medicine, University of Tunis El Manar, Tunis, Tunisia b Pediatric Immuno-Hematology Unit, Bone Marrow Transplantation Center Tunis, Tunis, Tunisia c Information System Directions, Sahloul University Hospital, Sousse, Tunisia d National Center of Blood Transfusion, Tunis, Tunisia ARTICLE INFO ABSTRACT Editor: Mohandas Narla In beta-thalassemia patients, erythrocyte autoantibodies can remain silent or lead to Autoimmune Hemolytic Keywords: Anemia (AIHA).The aim of this study was to identify predictors of AIHA in beta-thalassemia patients with Autoimmune hemolytic anemia positive Direct Antiglobulin Test (DAT), in Tunisia. Thalassemia This longitudinal prognosis study was carried out on beta-thalassemia patients with a positive confirmed Transfusion DAT. Predictors of AIHA were identified the Kaplan-Meier method. A Cox model analysis was used to identify Autoantibodies independent predictors. Red blood cells Among 385 beta thalassemia patients, 87 developed positive DAT (22.6%). Autoimmune hemolytic anemia Direct antiglobulin test was occurred in 25 patients. Multivariate analysis showed that AIHA was independently associated with beta- thalassemia intermedia and similar family history of AIHA. Splenectomy in patients with positive DAT was independently associated with an increased risk of AIHA (HR = 6.175, CI: 2.049–18.612, p < 0.001). -

Hb S/Beta-Thalassemia
rev bras hematol hemoter. 2 0 1 5;3 7(3):150–152 Revista Brasileira de Hematologia e Hemoterapia Brazilian Journal of Hematology and Hemotherapy www.rbhh.org Scientific Comment ଝ The compound state: Hb S/beta-thalassemia ∗ Maria Stella Figueiredo Universidade Federal de São Paulo (UNIFESP), São Paulo, SP, Brazil Sickle cell disease (SCD) results from a single amino acid small increase in Hb concentration and in packed cell volume. substitution in the gene encoding the -globin subunit However, these effects are not accompanied by any reduction  ( 6Glu > Val) that produces the abnormal hemoglobin (Hb) in vaso-occlusive events, probably due to the great number of named Hb S. SCD has different genotypes with substantial Hb S-containing red blood cells resulting in increased blood 1,2 4,5 variations in presentation and clinical course (Table 1). The viscosity. combination of the sickle cell mutation and beta-thalassemia A confusing diagnostic problem is the differentiation 0  (-Thal) mutation gives rise to a compound heterozygous con- of Hb S/ -Thal from sickle cell anemia associated with ␣ dition known as Hb S/ thalassemia (Hb S/-Thal), which was -thalassemia. Table 2 shows that hematologic and elec- 3 first described in 1944 by Silvestroni and Bianco. trophoretic studies are unable to distinguish between the two The polymerization of deoxygenated Hb S (sickling) is the conditions and so family studies and DNA analysis are needed 5 primary event in the molecular pathogenesis of SCD. How- to confirm the diagnosis. +  ever, this event is highly dependent on the intracellular Hb In Hb S/ -Thal, variable amounts of Hb A dilute Hb S and composition; in other words, it is dependent on the concen- consequently inhibit polymerization-induced cellular dam- tration of Hb S, and type and concentration of the other types age. -

Non-Transfusion-Dependent Thalassemias
REVIEW ARTICLES Non-transfusion-dependent thalassemias Khaled M. Musallam, 1 Stefano Rivella, 2 Elliott Vichinsky, 3 and Eliezer A. Rachmilewitz, 4 1Department of Medicine and Medical Specialties, IRCCS Ca’ Granda Foundation Maggiore Policlinico Hospital, University of Milan, Milan, Italy; 2Department of Pediatrics, Division of Hematology/Oncology, Weill Medical College of Cornell University, New York, NY, USA; 3Department of Hematology and Oncology, Children’s Hospital and Research Center Oakland, Oakland, CA, USA; and 4Department of Hematology, Wolfson Medical Center, Holon, Israel ABSTRACT Non-transfusion-dependent thalassemias include a variety of phenotypes that, unlike patients with beta ( β)-tha - lassemia major, do not require regular transfusion therapy for survival. The most commonly investigated forms are β-thalassemia intermedia, hemoglobin E/ β-thalassemia, and α-thalassemia intermedia (hemoglobin H disease). However, transfusion-independence in such patients is not without side effects. Ineffective erythropoiesis and peripheral hemolysis, the hallmarks of disease process, lead to a variety of subsequent pathophysiologies including iron overload and hypercoagulability that ultimately lead to a number of serious clinical morbidities. Thus, prompt and accurate diagnosis of non-transfusion-dependent thalassemia is essential to ensure early intervention. Although several management options are currently available, the need to develop more novel therapeutics is jus - tified by recent advances in our understanding of the mechanisms of disease. Such efforts require wide interna - tional collaboration, especially since non-transfusion-dependent thalassemias are no longer bound to low- and middle-income countries but have spread to large multiethnic cities in Europe and the Americas due to continued migration. Introduction survival, although they may require occasional or even fre - quent transfusions in certain clinical settings and usually for Inherited hemoglobin disorders can be divided into two defined periods of time (Figure 1). -

The Variable Manifestations of Disease in Pyruvate Kinase Deficiency and Their Management
REVIEW ARTICLE The variable manifestations of disease in pyruvate kinase deficiency and their Ferrata Storti Foundation management Hanny Al-Samkari,1 Eduard J. van Beers,2 Kevin H.M. Kuo,3 Wilma Barcellini,4 Paola Bianchi,4 Andreas Glenthøj,5 María del Mar Mañú Pereira,6 Richard van Wijk,7 Bertil Glader8 and Rachael F. Grace9 1Division of Hematology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; 2Van Creveldkliniek, University Medical Centre Utrecht, University of Utrecht, Utrecht, the Netherlands; 3Division of Hematology, University of Toronto, University Health Haematologica 2020 Network, Toronto, Ontario, Canada; 4UOS Ematologia, Fisiopatologia delle Anemie, Volume 105(9):2229-2239 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy; 5Department of Hematology, Herlev and Gentofte Hospital, Herlev, Denmark; 6Translational Research in Rare Anaemia Disorders, Vall d'Hebron Institut de Recerca (VHIR), Barcelona, Spain; 7Department of Clinical Chemistry & Hematology, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands; 8Lucile Packard Children’s Hospital, Stanford University School of Medicine, Palo Alto, CA, USA and 9Dana/Farber Boston Children's Cancer and Blood Disorders Center, Harvard Medical School, Boston, MA, USA. ABSTRACT yruvate kinase deficiency (PKD) is the most common cause of chron- ic hereditary non-spherocytic hemolytic anemia and results in a Pbroad spectrum of disease. The diagnosis of PKD requires a high index of suspicion and judicious use of laboratory tests that may not always be informative, including pyruvate kinase enzyme assay and genetic analysis of the PKLR gene. A significant minority of patients with PKD have occult mutations in non-coding regions of PKLR which are missed on standard genetic tests. -

The Hematological Complications of Alcoholism
The Hematological Complications of Alcoholism HAROLD S. BALLARD, M.D. Alcohol has numerous adverse effects on the various types of blood cells and their functions. For example, heavy alcohol consumption can cause generalized suppression of blood cell production and the production of structurally abnormal blood cell precursors that cannot mature into functional cells. Alcoholics frequently have defective red blood cells that are destroyed prematurely, possibly resulting in anemia. Alcohol also interferes with the production and function of white blood cells, especially those that defend the body against invading bacteria. Consequently, alcoholics frequently suffer from bacterial infections. Finally, alcohol adversely affects the platelets and other components of the blood-clotting system. Heavy alcohol consumption thus may increase the drinker’s risk of suffering a stroke. KEY WORDS: adverse drug effect; AODE (alcohol and other drug effects); blood function; cell growth and differentiation; erythrocytes; leukocytes; platelets; plasma proteins; bone marrow; anemia; blood coagulation; thrombocytopenia; fibrinolysis; macrophage; monocyte; stroke; bacterial disease; literature review eople who abuse alcohol1 are at both direct and indirect. The direct in the number and function of WBC’s risk for numerous alcohol-related consequences of excessive alcohol increases the drinker’s risk of serious Pmedical complications, includ- consumption include toxic effects on infection, and impaired platelet produc- ing those affecting the blood (i.e., the the bone marrow; the blood cell pre- tion and function interfere with blood cursors; and the mature red blood blood cells as well as proteins present clotting, leading to symptoms ranging in the blood plasma) and the bone cells (RBC’s), white blood cells from a simple nosebleed to bleeding in marrow, where the blood cells are (WBC’s), and platelets. -

Concurrent Sickle Cell Anemia and Alpha-Thalassemia. Effect on Pathological Properties of Sickle Erythrocytes
Concurrent sickle cell anemia and alpha-thalassemia. Effect on pathological properties of sickle erythrocytes. S H Embury, … , G Monroy, N Mohandas J Clin Invest. 1984;73(1):116-123. https://doi.org/10.1172/JCI111181. Research Article The concurrence of sickle cell anemia and alpha-thalassemia results in less severe hemolytic anemia apparently as a result of reduced intraerythrocytic concentration of hemoglobin S and its retarded polymerization. We have evaluated the effect of alpha-globin gene number on several interrelated properties of sickle erythrocytes (RBC) that are expected to correlate with the hemolytic and rheologic consequences of sickle cell disease. The irreversibly sickled cell number, proportion of very dense sickle RBC, and diminished deformability of sickle RBC each varied directly with alpha-globin gene number. Sickle RBC density was a direct function of the mean corpuscular hemoglobin concentration (MCHC). Even in nonsickle RBC, alpha-globin gene number varied directly with RBC density. Despite differences in alpha-globin gene number, sickle RBC of the same density had the same degree of deformability and dehydration. These data indicate that the fundamental effect of alpha-thalassemia is to inhibit the generation of sickle RBC having high density and MCHC, and that the other beneficial effects of sickle RBC are secondary to this process. The less consistent effect on overall clinical severity reported for subjects with this concurrence may reflect an undefined detrimental effect of alpha-thalassemia, possibly on the whole blood viscosity or on sickle RBC membrane-mediated adherence phenomena. Find the latest version: https://jci.me/111181/pdf Concurrent Sickle Cell Anemia and a-Thalassemia Effect on Pathological Properties of Sickle Erythrocytes Stephen H. -

Hemoglobin Wayne Trait with Incidental Polycythemia
Available online at www.annclinlabsci.org 96 Annals of Clinical & Laboratory Science, vol. 47, no. 1, 2017 Hemoglobin Wayne Trait with Incidental Polycythemia Manju Ambelil, Nghia Nguyen, Amitava Dasgupta, Semyon Risin, and Amer Wahed Department of Pathology and Laboratory Medicine, University of Texas Health Science Center- McGovern Medical School, Houston, TX, USA Abstract. Hemoglobinopathies, caused by mutations in the globin genes, are one of the most common inherited disorders. Many of the hemoglobin variants can be identified by hemoglobin analysis using conventional electrophoresis and high performance liquid chromatography; however hemoglobin DNA analysis may be necessary in other cases for confirmation. Here, we report a case of a rare alpha chain he- moglobin variant, hemoglobin Wayne, in a 47-year-old man who presented with secondary polycythemia. Capillary zone electrophoresis and high performance liquid chromatography revealed a significant amount of a hemoglobin variant, which was further confirmed by hemoglobin DNA sequencing as hemoglobin Wayne. Since the patient was not homozygous for hemoglobin Wayne, which is associated with second- ary polycythemia, the laboratory diagnosis in this case was critical in ruling out hemoglobinopathy as the etiology of his polycythemia. Key words: Hemoglobinopathies; hemoglobin Wayne; alpha globin chain variant. Introduction mean corpuscular hemoglobin of 27.1 pg. The red cell distribution width, total and differential leukocyte Hemoglobinopathies are the most common and counts, and platelets were normal. An anemia study clinically significant single-gene-inherited disorders showed a serum iron level of 127 mcg/dL, a ferritin con- [1]. These are caused by mutations in the globin centration of 239 ng/mL, an iron saturation of 42%, a total iron binding capacity of 306 mcg/dL, an unsatu- genes, resulting in quantitative and qualitative de- rated iron binding capacity of 179mcg/dL, and a vita- fects in the globin chain synthesis [2]. -

Iron Deficiency and the Anemia of Chronic Disease
Thomas G. DeLoughery, MD MACP FAWM Professor of Medicine, Pathology, and Pediatrics Oregon Health Sciences University Portland, Oregon [email protected] IRON DEFICIENCY AND THE ANEMIA OF CHRONIC DISEASE SIGNIFICANCE Lack of iron and the anemia of chronic disease are the most common causes of anemia in the world. The majority of pre-menopausal women will have some element of iron deficiency. The first clue to many GI cancers and other diseases is iron loss. Finally, iron deficiency is one of the most treatable medical disorders of the elderly. IRON METABOLISM It is crucial to understand normal iron metabolism to understand iron deficiency and the anemia of chronic disease. Iron in food is largely in ferric form (Fe+++ ) which is reduced by stomach acid to the ferrous form (Fe++). In the jejunum two receptors on the mucosal cells absorb iron. The one for heme-iron (heme iron receptor) is very avid for heme-bound iron (absorbs 30-40%). The other receptor - divalent metal transporter (DMT1) - takes up inorganic iron but is less efficient (1-10%). Iron is exported from the enterocyte via ferroportin and is then delivered to the transferrin receptor (TfR) and then to plasma transferrin. Transferrin is the main transport molecule for iron. Transferrin can deliver iron to the marrow for the use in RBC production or to the liver for storage in ferritin. Transferrin binds to the TfR on the cell and iron is delivered either for use in hemoglobin synthesis or storage. Iron that is contained in hemoglobin in senescent red cells is recycled by binding to ferritin in the macrophage and is transferred to transferrin for recycling. -

Case Report on Methylene Blue Induced Hemolytic Anemia
Available online at www.ijmrhs.com al R edic ese M a of rc l h a & n r H u e o a J l l t h International Journal of Medical Research & a n S ISSN No: 2319-5886 o c i t i Health Sciences, 2019, 8(5): 83-85 e a n n c r e e t s n I • • I J M R H S Case Report on Methylene Blue Induced Hemolytic Anemia Sulfath T.S1, Bhanu Kumar M2, Koneru Vasavi1, Aparna R Menon1 and Ann V Kuruvilla1* 1 Department of Pharmacy Practice, JSS College of Pharmacy, Mysuru Jagadguru Shri Shivarathreeshwara Academy of Higher Education and Research, Karnataka, India 2 Department of General Medicine, JSS Medical College and Hospital, JSS Academy of Higher Education and Research, Karnataka, India *Corresponding e-mail: [email protected] ABSTRACT A 22-years old male patient was admitted to a tertiary care hospital with complaints of an alleged history of intentional poisoning (organophosphorus compound and nitrofurantoin) and developed hemolytic anemia after receiving methylene blue for 8 days. The patient presented with hematuria and hemoglobin level 3.1 which confirmed hemolytic anemia G6PD level was normal. Methylene blue was discontinued and PRBC transfusion (3 pints) was given. After 4 days of blood transfusion, the patient’s Hb level became 9.4 g/dl. Causality assessment was suggestive of a probable relationship between the drug and reaction. Keywords: Hemolytic anemia, Methylene blue, G6PD Abbreviations: G6PD: Glucose-6-phosphate Dehydrogenase; PRBC: Packed Red Blood Cells; LMB: Leucomethylene Blue; NADPH: Nicotinamide Adenine Dinucleotide Phosphate; OP: Organophosphorus; SPO2: Saturation of peripheral oxygen; CBC: Complete Blood Count INTRODUCTION Methylene blue (tetramethylthionine chloride) is a diagnostic agent in kidney function, anti-infective agent, antidote, antiseptic and nutraceutical. -
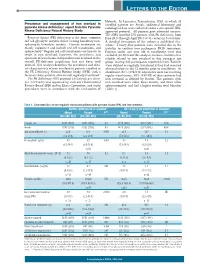
Prevalence and Management of Iron Overload in Pyruvate Kinase Deficiency
LETTERS TO THE EDITOR Helsinki. In Lancaster, Pennsylvania, USA, in which all Prevalence and management of iron overload in enrolled patients are Amish, additional laboratory and pyruvate kinase deficiency: report from the Pyruvate radiological data were collected under a site-specific IRB- Kinase Deficiency Natural History Study approved protocol. All patients gave informed consent. The NHS enrolled 278 patients with PK deficiency from Pyruvate kinase (PK) deficiency is the most common June 2014 through April 2017 at 31 centers in 6 countries. red cell glycolytic enzyme defect causing hereditary non- A detailed description of the cohort is published else- spherocytic hemolytic anemia. Current treatments are where.2 Twenty-four patients were excluded due to the mainly supportive and include red cell transfusions and inability to confirm two pathogenic PKLR mutations. splenectomy.1 Regular red cell transfusions are known to Patients under one year old at enrollment were also result in iron overload; however, the prevalence and excluded (n=12) from this analysis, because ferritin is less spectrum of transfusion-independent iron overload in the reliably related to iron overload in this youngest age overall PK-deficient population has not been well group, leaving 242 participants reported herein. Patients defined. This analysis describes the prevalence and clini- were defined as regularly transfused if they had received cal characteristics of iron overload in patients enrolled in ≥6 transfusions in the 12 months prior to enrollment. At the PK Deficiency Natural History Study (NHS) with a enrollment, 82% (198/242) of patients were not receiving focus on those patients who are not regularly transfused.2 regular transfusions; 38% (53/138) of these patients had The PK deficiency NHS protocol (clinicaltrials.gov identi- iron overload as defined by ferritin.