Regulation by Non-Coding Rnas Volume 2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Expression of NOD2, NLRP3 and NLRC5 and Renal Injury in Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis
Wang et al. J Transl Med (2019) 17:197 https://doi.org/10.1186/s12967-019-1949-5 Journal of Translational Medicine RESEARCH Open Access The expression of NOD2, NLRP3 and NLRC5 and renal injury in anti-neutrophil cytoplasmic antibody-associated vasculitis Luo‑Yi Wang1,2,3, Xiao‑Jing Sun1,2,3, Min Chen1,2,3* and Ming‑Hui Zhao1,2,3,4 Abstract Background: Nucleotide‑binding oligomerization domain (NOD)‑like receptors (NLRs) are intracellular sensors of pathogens and molecules from damaged cells to regulate the infammatory response in the innate immune system. Emerging evidences suggested a potential role of NLRs in anti‑neutrophil cytoplasmic antibody (ANCA)‑associated vasculitis (AAV). This study aimed to investigate the expression of nucleotide‑binding oligomerization domain con‑ taining protein 2 (NOD2), NOD‑like receptor family pyrin domain containing 3 (NLRP3) and NOD‑like receptor family CARD domain containing 5 (NLRC5) in kidneys of AAV patients, and further explored their associations with clinical and pathological parameters. Methods: Thirty‑four AAV patients in active stage were recruited. Their renal specimens were processed with immu‑ nohistochemistry to assess the expression of three NLRs, and with double immunofuorescence to detect NLRs on intrinsic and infltrating cells. Analysis of gene expression was also adopted in cultured human podocytes. The associa‑ tions between expression of NLRs and clinicopathological parameters were analyzed. Results: The expression of NOD2, NLRP3 and NLRC5 was signifcantly higher in kidneys from AAV patients than those from normal controls, minimal change disease or class IV lupus nephritis. These NLRs co‑localized with podocytes and infltrating infammatory cells. -
Eukaryotic Non-Coding Rnas: New Targets for Diagnostics and Therapeutics?
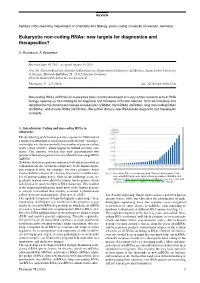
REVIEW Institute of Biochemistry, Department of Chemistry and Biology, Justus Liebig University of Giessen, Germany Eukaryotic non-coding RNAs: new targets for diagnostics and therapeutics? O. Rossbach, A. Bindereif Received June 30, 2015, accepted August 19, 2015 Prof. Dr. Albrecht Bindereif, Institute of Biochemistry, Department of Chemistry and Biology, Justus Liebig University of Giessen, Heinrich-Buff-Ring 58, 35392 Giessen, Germany [email protected] Pharmazie 71: 3–7 (2016) doi: 10.1691/ph.2016.5736 Non-coding RNAs (ncRNAs) in eukaryotes have recently developed to a very active research area in RNA biology, opening up new strategies for diagnosis and therapies of human disease. Here we introduce and describe the most important classes of eukaryotic ncRNAs: microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs). We further discuss new RNA-based diagnostic and therapeutic concepts. 1. Introduction: Coding and non-coding RNAs in eukaryotes The deciphering of the human genome sequence in 2000 marked a unique breakthrough in modern molecular biology. An impor- tant insight was the unexpectedly low number of protein-coding genes (about 20,000), which lagged far behind previous esti- mates. This number revealed that only approximately two percent of the human genome is transcribed into messenger RNA (mRNA). However, the human genome sequence itself did not provide an explanation for the enormous complexity of the human organ- ism compared with, for example, the more primitive worm Caenorhabditis elegans (C. elegans) that carries a similar num- Fig. 1: Non-coding RNAs as an emerging field. The rapid development of the ber of protein-coding genes. -

The ELIXIR Core Data Resources: Fundamental Infrastructure for The
Supplementary Data: The ELIXIR Core Data Resources: fundamental infrastructure for the life sciences The “Supporting Material” referred to within this Supplementary Data can be found in the Supporting.Material.CDR.infrastructure file, DOI: 10.5281/zenodo.2625247 (https://zenodo.org/record/2625247). Figure 1. Scale of the Core Data Resources Table S1. Data from which Figure 1 is derived: Year 2013 2014 2015 2016 2017 Data entries 765881651 997794559 1726529931 1853429002 2715599247 Monthly user/IP addresses 1700660 2109586 2413724 2502617 2867265 FTEs 270 292.65 295.65 289.7 311.2 Figure 1 includes data from the following Core Data Resources: ArrayExpress, BRENDA, CATH, ChEBI, ChEMBL, EGA, ENA, Ensembl, Ensembl Genomes, EuropePMC, HPA, IntAct /MINT , InterPro, PDBe, PRIDE, SILVA, STRING, UniProt ● Note that Ensembl’s compute infrastructure physically relocated in 2016, so “Users/IP address” data are not available for that year. In this case, the 2015 numbers were rolled forward to 2016. ● Note that STRING makes only minor releases in 2014 and 2016, in that the interactions are re-computed, but the number of “Data entries” remains unchanged. The major releases that change the number of “Data entries” happened in 2013 and 2015. So, for “Data entries” , the number for 2013 was rolled forward to 2014, and the number for 2015 was rolled forward to 2016. The ELIXIR Core Data Resources: fundamental infrastructure for the life sciences 1 Figure 2: Usage of Core Data Resources in research The following steps were taken: 1. API calls were run on open access full text articles in Europe PMC to identify articles that mention Core Data Resource by name or include specific data record accession numbers. -

Striking the Right Balance in Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis
Striking the Right Balance in Anti-neutrophil Cytoplasmic Antibody-Associated Vasculitis This symposium took place on 4th June 2021, as part of the European Alliance of Associations for Rheumatology (EULAR) virtual congress Speakers: Benjamin Terrier,¹ Joanna Robson,² Bernhard Hellmich³ 1. University of Paris and Hôpital Cochin, France 2. University of the West of England and Bristol Royal Infirmary, UK 3. University of Tübingen, Germany Disclosure: Terrier has been an advisory board member and/or received consulting fees/ travel expenses from AstraZeneca, Chugai, Grifols, GlaxoSmithKline, Janssen, LFB, Octapharma, Roche, and Vifor Pharma. Robson has received speaker’s fees from Roche and Vifor Pharma; and research support from Vifor Pharma. Hellmich has been an investigator in clinical trials for Ab2Bio, AbbVie, AstraZeneca, Bristol- Myers Squibb, Chemocentrix, GlaxoSmithKline, InflaRx, Kiniksa, Nippon Kayaku, Novartis, Roche, and Sanofi. He has acted as a consultant, advisory board member, and/or lecturer for AbbVie, Bristol-Myers Squibb, Boehringer Ingelheim, Chugai, GlaxoSmithKline, InflaRx, Novartis, Pfizer, Roche, and Vifor Pharma. He is also a member of the Guideline Committees for European Alliance of Associations for Rheumatology (EULAR) and the German Society of Rheumatology (DGRh). Acknowledgements: Writing assistance was provided by Helen Boreham. Support: The publication of this article was funded by Vifor Pharma. The views and opinions expressed are those of the presenters. Content was reviewed by Vifor Pharma for medical accuracy. Citation: Rheumatol. 2021;8[1]:43-50. Meeting Summary Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) causes irreversible short- and long-term damage to vital organs, particularly the kidneys and lungs. Current standard of care (SOC) for AAV, of which glucocorticoids (GC) are a lynchpin, has a number of important limitations: responses to therapy are variable, some patients fail to achieve and sustain remission, and treatment related adverse events (AE) are common. -

Integrative Differential Expression and Gene Set Enrichment Analysis Using Summary Statistics for Scrna-Seq Studies
ARTICLE https://doi.org/10.1038/s41467-020-15298-6 OPEN Integrative differential expression and gene set enrichment analysis using summary statistics for scRNA-seq studies ✉ Ying Ma 1,7, Shiquan Sun 1,7, Xuequn Shang2, Evan T. Keller 3, Mengjie Chen 4,5 & Xiang Zhou 1,6 Differential expression (DE) analysis and gene set enrichment (GSE) analysis are commonly applied in single cell RNA sequencing (scRNA-seq) studies. Here, we develop an integrative 1234567890():,; and scalable computational method, iDEA, to perform joint DE and GSE analysis through a hierarchical Bayesian framework. By integrating DE and GSE analyses, iDEA can improve the power and consistency of DE analysis and the accuracy of GSE analysis. Importantly, iDEA uses only DE summary statistics as input, enabling effective data modeling through com- plementing and pairing with various existing DE methods. We illustrate the benefits of iDEA with extensive simulations. We also apply iDEA to analyze three scRNA-seq data sets, where iDEA achieves up to five-fold power gain over existing GSE methods and up to 64% power gain over existing DE methods. The power gain brought by iDEA allows us to identify many pathways that would not be identified by existing approaches in these data. 1 Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA. 2 School of Computer Science, Northwestern Polytechnical University, Xi’an, Shaanxi 710072, P.R. China. 3 Department of Urology, University of Michigan, Ann Arbor, MI 48109, USA. 4 Department of Human Genetics, University of Chicago, Chicago, IL 60637, USA. 5 Section of Genetic Medicine, Department of Medicine, University of Chicago, Chicago, IL 60637, USA. -
Lymphocyte Separation Medium (LSM
THE JOURNAL OF IMMUNOLOGY Bionetics does it for you. Lymphocyte Separation Medium (LSM wenient One-Step Centrifugation Method _ayer diluted blood on LSM. 2,entrifuge for 30-40 min., 18-20°C, ~.00 x g. ~,spirate and discard plasma layer -larvest lymphocyte layer. Quality Control Assurance Each lot is tested for: • Lymphocyte separation and recovery. • Lymphocyte viability. • Sterility. • Consistent density (1.077-1.080 at 20°C). Packaging • Packaged in amber, screw-cap bottles. • 5 x 100 ml bottles per carton. Storage. • Stored at room temperature. Reference Boyum, A. (1968): Isolation of mononuclear cells and granulocytes from human blood. Scand J. Clin. Lab. Invest. 21, Suppl. 97. Aspirate I IC[OI I IC~ LJI Catalog number: 8410-01 & discard serum Lymphocyte For Laboratory Use Aspirate layer & use (mononuclear Please write for our current Price List and Catalog. cells and Original platelets) ITi BIONETICS° LSM layer Erythrocytes Laboratory Products and Litton granulocytes 5516 Nicholson Lane, Kensington, Maryland 20795 Telephone: (301) 881-1557 1979 Litton Bionetics, tnc Get the most out of your high quality cytotoxic antibodies with LOW-TOX-M RABBIT COMPLEMENT LOW TOXICITY HIGH ACTIVITY Presentation: CL 3051 5 x 1 ml, lyophilized $30.00 When it comes to COMPLEMENT... come to CEDARLANE Direct orders or inquiries to: UNITED STATES: WORLDWIDE EXCEPT U.S. ,4 C~L CEDARLANE ACCURATE CHEMICAL & LABORATO RI ES SCIENTIFIC CORPORATION LIMITED 5516-8TH LINE, R.R. 2 28 TEC STREET, HICKSVtLLE, N.Y. 11801 HORNBY, ONTARIO, CANADA LOP 1E0 Telephone -

Beet Curly Top Virus Strains Associated with Sugar Beet in Idaho, Oregon, and a Western U.S
Plant Disease • 2017 • 101:1373-1382 • http://dx.doi.org/10.1094/PDIS-03-17-0381-RE Beet curly top virus Strains Associated with Sugar Beet in Idaho, Oregon, and a Western U.S. Collection Carl A. Strausbaugh and Imad A. Eujayl, United States Department of Agriculture–Agricultural Research Service (USDA-ARS) Northwest Irrigation and Soils Research Laboratory, Kimberly, ID 83341; and William M. Wintermantel, USDA-ARS, Salinas, CA 93905 Abstract Curly top of sugar beet is a serious, yield-limiting disease in semiarid pro- Logan) strains and primers that amplified a group of Worland (Wor)- duction areas caused by Beet curly top virus (BCTV) and transmitted like strains. The BCTV strain distribution averaged 2% Svr, 30% CA/ by the beet leafhopper. One of the primary means of control for BCTV Logan, and 87% Wor-like (16% had mixed infections), which differed in sugar beet is host resistance but effectiveness of resistance can vary from the previously published 2006-to-2007 collection (87% Svr, 7% among BCTV strains. Strain prevalence among BCTV populations CA/Logan, and 60% Wor-like; 59% mixed infections) based on a contin- was last investigated in Idaho and Oregon during a 2006-to-2007 collec- gency test (P < 0.0001). Whole-genome sequencing (GenBank acces- tion but changes in disease severity suggested a need for reevaluation. sions KT276895 to KT276920 and KX867015 to KX867057) with Therefore, 406 leaf samples symptomatic for curly top were collected overlapping primers found that the Wor-like strains included Wor, Colo- from sugar beet plants in commercial sugar beet fields in Idaho and rado and a previously undescribed strain designated Kimberly1. -

Methods in and Applications of the Sequencing of Short Non-Coding Rnas" (2013)
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2013 Methods in and Applications of the Sequencing of Short Non- Coding RNAs Paul Ryvkin University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, Genetics Commons, and the Molecular Biology Commons Recommended Citation Ryvkin, Paul, "Methods in and Applications of the Sequencing of Short Non-Coding RNAs" (2013). Publicly Accessible Penn Dissertations. 922. https://repository.upenn.edu/edissertations/922 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/922 For more information, please contact [email protected]. Methods in and Applications of the Sequencing of Short Non-Coding RNAs Abstract Short non-coding RNAs are important for all domains of life. With the advent of modern molecular biology their applicability to medicine has become apparent in settings ranging from diagonistic biomarkers to therapeutics and fields angingr from oncology to neurology. In addition, a critical, recent technological development is high-throughput sequencing of nucleic acids. The convergence of modern biotechnology with developments in RNA biology presents opportunities in both basic research and medical settings. Here I present two novel methods for leveraging high-throughput sequencing in the study of short non- coding RNAs, as well as a study in which they are applied to Alzheimer's Disease (AD). The computational methods presented here include High-throughput Annotation of Modified Ribonucleotides (HAMR), which enables researchers to detect post-transcriptional covalent modifications ot RNAs in a high-throughput manner. In addition, I describe Classification of RNAs by Analysis of Length (CoRAL), a computational method that allows researchers to characterize the pathways responsible for short non-coding RNA biogenesis. -

A Pirna-Like Small RNA Interacts with and Modulates P-ERM Proteins in Human Somatic Cells
ARTICLE Received 18 Mar 2015 | Accepted 27 Apr 2015 | Published 22 June 2015 DOI: 10.1038/ncomms8316 OPEN A piRNA-like small RNA interacts with and modulates p-ERM proteins in human somatic cells Yuping Mei1, Yuyan Wang1,2, Priti Kumari3, Amol Carl Shetty3, David Clark1, Tyler Gable1, Alexander D. MacKerell4,5, Mark Z. Ma1, David J. Weber5,6, Austin J. Yang5, Martin J. Edelman5 & Li Mao1,5 PIWI-interacting RNAs (piRNAs) are thought to silence transposon and gene expression during development. However, the roles of piRNAs in somatic tissues are largely unknown. Here we report the identification of 555 piRNAs in human lung bronchial epithelial (HBE) and non-small cell lung cancer (NSCLC) cell lines, including 295 that do not exist in databases termed as piRNA-like sncRNAs or piRNA-Ls. Distinctive piRNA/piRNA-L expression patterns are observed between HBE and NSCLC cells. piRNA-like-163 (piR-L-163), the top down- regulated piRNA-L in NSCLC cells, binds directly to phosphorylated ERM proteins (p-ERM), which is dependent on the central part of UUNNUUUNNUU motif in piR-L-163 and the RRRKPDT element in ERM. The piR-L-163/p-ERM interaction is critical for p-ERM’s binding capability to filamentous actin (F-actin) and ERM-binding phosphoprotein 50 (EBP50). Thus, piRNA/piRNA-L may play a regulatory role through direct interaction with proteins in physiological and pathophysiological conditions. 1 Department of Oncology and Diagnostic Sciences, University of Maryland School of Dentistry, 650W Baltimore Street, Baltimore, Maryland 21201, USA. 2 Department of Thoracic Medical Oncology, Peking University Cancer Hospital and Institute, Beijing 100142, China. -

Comparing Tools for Non-Coding RNA Multiple Sequence Alignment Based On
Downloaded from rnajournal.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press ES Wright 1 1 TITLE 2 RNAconTest: Comparing tools for non-coding RNA multiple sequence alignment based on 3 structural consistency 4 Running title: RNAconTest: benchmarking comparative RNA programs 5 Author: Erik S. Wright1,* 6 1 Department of Biomedical Informatics, University of Pittsburgh (Pittsburgh, PA) 7 * Corresponding author: Erik S. Wright ([email protected]) 8 Keywords: Multiple sequence alignment, Secondary structure prediction, Benchmark, non- 9 coding RNA, Consensus secondary structure 10 Downloaded from rnajournal.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press ES Wright 2 11 ABSTRACT 12 The importance of non-coding RNA sequences has become increasingly clear over the past 13 decade. New RNA families are often detected and analyzed using comparative methods based on 14 multiple sequence alignments. Accordingly, a number of programs have been developed for 15 aligning and deriving secondary structures from sets of RNA sequences. Yet, the best tools for 16 these tasks remain unclear because existing benchmarks contain too few sequences belonging to 17 only a small number of RNA families. RNAconTest (RNA consistency test) is a new 18 benchmarking approach relying on the observation that secondary structure is often conserved 19 across highly divergent RNA sequences from the same family. RNAconTest scores multiple 20 sequence alignments based on the level of consistency among known secondary structures 21 belonging to reference sequences in their output alignment. Similarly, consensus secondary 22 structure predictions are scored according to their agreement with one or more known structures 23 in a family. -

Induction of Protein Citrullination and Auto-Antibodies Production In
www.nature.com/scientificreports OPEN Induction of protein citrullination and auto-antibodies production in murine exposed to nickel Received: 11 November 2015 Accepted: 21 December 2017 nanomaterials Published: xx xx xxxx Bashir M. Mohamed1,7, Noreen T. Boyle2,3, Anja Schinwald4, Bruno Murer5, Ronan Ward6, Omar K. Mahfoud1, Tatsiana Rakovich1, Kieran Crosbie-Staunton1, Steven G. Gray 7, Ken Donaldson4, Yuri Volkov1,2 & Adriele Prina-Mello 1,2 Citrullination, or the post-translational deimination of polypeptide-bound arginine, is involved in several pathological processes in the body, including autoimmunity and tumorigenesis. Recent studies have shown that nanomaterials can trigger protein citrullination, which might constitute a common pathogenic link to disease development. Here we demonstrated auto-antibody production in serum of nanomaterials-treated mice. Citrullination-associated phenomena and PAD levels were found to be elevated in nanomaterials -treated cell lines as well as in the spleen, kidneys and lymph nodes of mice, suggesting a systemic response to nanomaterials injection, and validated in human pleural and pericardial malignant mesothelioma (MM) samples. The observed systemic responses in mice exposed to nanomaterials support the evidence linking exposure to environmental factors with the development of autoimmunity responses and reinforces the need for comprehensive safety screening of nanomaterials. Furthermore, these nanomaterials induce pathological processes that mimic those observed in Pleural MM, and therefore require further investigations into their carcinogenicity. Citrullination is involved in several pathological processes in the body, including autoimmunity and tumor- igenesis. Citrullinated proteins are generated by a post-translational deimination or demethylimination of polypeptide-bound arginine by a family of Ca2+-dependent enzyme peptidylarginine deiminase (PAD)1. -

Annual Scientific Report 2013 on the Cover Structure 3Fof in the Protein Data Bank, Determined by Laponogov, I
EMBL-European Bioinformatics Institute Annual Scientific Report 2013 On the cover Structure 3fof in the Protein Data Bank, determined by Laponogov, I. et al. (2009) Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nature Structural & Molecular Biology 16, 667-669. © 2014 European Molecular Biology Laboratory This publication was produced by the External Relations team at the European Bioinformatics Institute (EMBL-EBI) A digital version of the brochure can be found at www.ebi.ac.uk/about/brochures For more information about EMBL-EBI please contact: [email protected] Contents Introduction & overview 3 Services 8 Genes, genomes and variation 8 Molecular atlas 12 Proteins and protein families 14 Molecular and cellular structures 18 Chemical biology 20 Molecular systems 22 Cross-domain tools and resources 24 Research 26 Support 32 ELIXIR 36 Facts and figures 38 Funding & resource allocation 38 Growth of core resources 40 Collaborations 42 Our staff in 2013 44 Scientific advisory committees 46 Major database collaborations 50 Publications 52 Organisation of EMBL-EBI leadership 61 2013 EMBL-EBI Annual Scientific Report 1 Foreword Welcome to EMBL-EBI’s 2013 Annual Scientific Report. Here we look back on our major achievements during the year, reflecting on the delivery of our world-class services, research, training, industry collaboration and European coordination of life-science data. The past year has been one full of exciting changes, both scientifically and organisationally. We unveiled a new website that helps users explore our resources more seamlessly, saw the publication of ground-breaking work in data storage and synthetic biology, joined the global alliance for global health, built important new relationships with our partners in industry and celebrated the launch of ELIXIR.