Dottorato Di Ricerca International Doctorate in Structural Biology
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -

Marine Organisms: a Source of Biomedically Relevant Metallo M1
Marine organisms : a source of biomedically relevant metallo M1, M2 and M17 exopeptidase inhibitors Isel Pascual Alonso, Laura Rivera Méndez, Fabiola Almeida, Mario Ernesto Valdés Tresano, Yarini Arrebola Sánchez, Aida Hernández-Zanuy, Luis Álvarez-Lajonchere, Dagmara Díaz, Belinda Sánchez, Isabelle Florent, et al. To cite this version: Isel Pascual Alonso, Laura Rivera Méndez, Fabiola Almeida, Mario Ernesto Valdés Tresano, Yarini Arrebola Sánchez, et al.. Marine organisms : a source of biomedically relevant metallo M1, M2 and M17 exopeptidase inhibitors. REVISTA CUBANA DE CIENCIAS BIOLÓGICAS, 2020, 8 (2), pp.1- 36. hal-02944434 HAL Id: hal-02944434 https://hal.archives-ouvertes.fr/hal-02944434 Submitted on 21 Sep 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. REVISTA CUBANA DE CIENCIAS BIOLÓGICAS http://www.rccb.uh.cu ARTÍCULO DE REVISIÓN Marine organisms: a source of biomedically relevant metallo M1, M2 and M17 exopeptidase inhibitors Los organismos marinos: Fuente de inhibidores de exopeptidasas de tipo metalo M1, M2 y M17 de relevancia biomédica Isel Pascual Alonso1 , Laura Rivera Méndez1 , Fabiola Almeida1 , Mario Ernesto Valdés Tresanco1,2 , Yarini Arrebola Sánchez1 , Aida Hernández-Zanuy3 , Luis Álvarez-Lajonchere4 , Dagmara Díaz1 , Belinda Sánchez5 , Isabelle Florent6 , Marjorie Schmitt7 , Miguel Cisneros8 , Jean Louis Charli8 1 Center for Protein Studies, Faculty of ABSTRACT Biology, University of Havana, Cuba. -

Specific Aminopeptidases of Indigenous Lactobacillus Brevis and Lactobacillus Plantarum
African Journal of Biotechnology Vol. 11(88), pp. 15438-15445, 1 November, 2012 Available online at http://www.academicjournals.org/AJB DOI: 10.5897/AJB12.703 ISSN 1684–5315 ©2012 Academic Journals Full Length Research Paper Specific aminopeptidases of indigenous Lactobacillus brevis and Lactobacillus plantarum BELKHEIR Khadidja*, ROUDJ Salima, ZADI KARAM Halima and KARAM Nour Eddine Laboratoire de Biologie des Microorganismes et Biotechnologie (LBMB). Université d’Oran, Algérie. Accepted 5 September, 2012 Lactic acid bacteria play an important role in milk coagulation and cheese ripening. To select strains showing interesting industrial features, two indigenous lactobacilli (Lactobacillus brevis and Lactobacillus plantarum) were studied for aminopeptidase activity. Cell and cells free extract were tested for leucyl aminopeptidase activity on the chromogenic leucyl-p-nitroanilide substrate. Intracellu- lar and membrane enzymes were solubilized with glycine /lyzozyme treatment then purified by ammonium sulphate precipitation followed by Sephadex G100 and diethylaminoethyl (DEAE) ions exchange chromatography’s separation. The molecular weight of denatured proteins was estimated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE). Effects of several parameters, pH, temperature, some ions and inhibitors on purified enzyme activity were studied. Cellular amino- peptidase activity was higher for CHTD 27 strain than BH14 strain. No aminopeptidase activity was noted in the cell free extract. The results of chromatography sephadex G100 combined to those of electrophoresis allowed suggesting a dimer structure for the native enzyme. The Lb CHTD27 purified enzyme showed maximal activity at pH 6.6 and at 40°C. This enzyme was partially inhibited by ethylenediamine acetic acid (EDTA) and Cu2+ ions but increased by Na2+ and Co2+ ions. -

Studies of Structure and Function of Tripeptidyl-Peptidase II
Till familj och vänner List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I. Eriksson, S.; Gutiérrez, O.A.; Bjerling, P.; Tomkinson, B. (2009) De- velopment, evaluation and application of tripeptidyl-peptidase II se- quence signatures. Archives of Biochemistry and Biophysics, 484(1):39-45 II. Lindås, A-C.; Eriksson, S.; Josza, E.; Tomkinson, B. (2008) Investiga- tion of a role for Glu-331 and Glu-305 in substrate binding of tripepti- dyl-peptidase II. Biochimica et Biophysica Acta, 1784(12):1899-1907 III. Eklund, S.; Lindås, A-C.; Hamnevik, E.; Widersten, M.; Tomkinson, B. Inter-species variation in the pH dependence of tripeptidyl- peptidase II. Manuscript IV. Eklund, S.; Kalbacher, H.; Tomkinson, B. Characterization of the endopeptidase activity of tripeptidyl-peptidase II. Manuscript Paper I and II were published under maiden name (Eriksson). Reprints were made with permission from the respective publishers. Contents Introduction ..................................................................................................... 9 Enzymes ..................................................................................................... 9 Enzymes and pH dependence .............................................................. 11 Peptidases ................................................................................................. 12 Serine peptidases ................................................................................. 14 Intracellular protein -

Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases
Handbook of Proteolytic Enzymes Second Edition Volume 1 Aspartic and Metallo Peptidases Alan J. Barrett Neil D. Rawlings J. Fred Woessner Editor biographies xxi Contributors xxiii Preface xxxi Introduction ' Abbreviations xxxvii ASPARTIC PEPTIDASES Introduction 1 Aspartic peptidases and their clans 3 2 Catalytic pathway of aspartic peptidases 12 Clan AA Family Al 3 Pepsin A 19 4 Pepsin B 28 5 Chymosin 29 6 Cathepsin E 33 7 Gastricsin 38 8 Cathepsin D 43 9 Napsin A 52 10 Renin 54 11 Mouse submandibular renin 62 12 Memapsin 1 64 13 Memapsin 2 66 14 Plasmepsins 70 15 Plasmepsin II 73 16 Tick heme-binding aspartic proteinase 76 17 Phytepsin 77 18 Nepenthesin 85 19 Saccharopepsin 87 20 Neurosporapepsin 90 21 Acrocylindropepsin 9 1 22 Aspergillopepsin I 92 23 Penicillopepsin 99 24 Endothiapepsin 104 25 Rhizopuspepsin 108 26 Mucorpepsin 11 1 27 Polyporopepsin 113 28 Candidapepsin 115 29 Candiparapsin 120 30 Canditropsin 123 31 Syncephapepsin 125 32 Barrierpepsin 126 33 Yapsin 1 128 34 Yapsin 2 132 35 Yapsin A 133 36 Pregnancy-associated glycoproteins 135 37 Pepsin F 137 38 Rhodotorulapepsin 139 39 Cladosporopepsin 140 40 Pycnoporopepsin 141 Family A2 and others 41 Human immunodeficiency virus 1 retropepsin 144 42 Human immunodeficiency virus 2 retropepsin 154 43 Simian immunodeficiency virus retropepsin 158 44 Equine infectious anemia virus retropepsin 160 45 Rous sarcoma virus retropepsin and avian myeloblastosis virus retropepsin 163 46 Human T-cell leukemia virus type I (HTLV-I) retropepsin 166 47 Bovine leukemia virus retropepsin 169 48 -

Product Sheet Info
Product Information Sheet for NR-19556 Salmonella enterica subsp. enterica, Strain supplemented with 15% glycerol. ® Ty2 (Serovar Typhi), Gateway Clone Set, Packaging/Storage: Recombinant in Escherichia coli, Plate 35 NR-19556 was packaged aseptically in a 96-well plate. The product is provided frozen and should be stored at -80°C or Catalog No. NR-19556 colder immediately upon arrival. For long-term storage, the This reagent is the tangible property of the U.S. Government. vapor phase of a liquid nitrogen freezer is recommended. Freeze-thaw cycles should be avoided. For research use only. Not for human use. Growth Conditions: Media: Contributor: LB broth or agar containing 50 µg/mL kanamycin. Pathogen Functional Genomics Resource Center at the J. Incubation: Craig Venter Institute Temperature: 37°C Atmosphere: Aerobic Manufacturer: Propagation: BEI Resources 1. Scrape top of frozen well with a pipette tip and streak onto agar plate. Product Description: 2. Incubate the plates at 37°C for 24 hours. Clone plates are replicated using a BioMek® FX robot. Production in the 96-well format has increased risk of cross- Citation: contamination between adjacent wells. Individual clones Acknowledgment for publications should read “The following should be purified (e.g. single colony isolation and reagent was obtained through BEI Resources, NIAID, purification using good microbiological practices) and NIH: Salmonella enterica subsp. enterica, Strain Ty2 sequence-verified prior to use. BEI Resources only confirms (Serovar Typhi), Gateway® Clone Set, Recombinant in the clone plate orientation and viability of randomly picked Escherichia coli, Plate 35, NR-19556.” clones. BEI Resources does not confirm or validate individual clone identities provided by the contributor. -

A Genomic Analysis of Rat Proteases and Protease Inhibitors
A genomic analysis of rat proteases and protease inhibitors Xose S. Puente and Carlos López-Otín Departamento de Bioquímica y Biología Molecular, Facultad de Medicina, Instituto Universitario de Oncología, Universidad de Oviedo, 33006-Oviedo, Spain Send correspondence to: Carlos López-Otín Departamento de Bioquímica y Biología Molecular Facultad de Medicina, Universidad de Oviedo 33006 Oviedo-SPAIN Tel. 34-985-104201; Fax: 34-985-103564 E-mail: [email protected] Proteases perform fundamental roles in multiple biological processes and are associated with a growing number of pathological conditions that involve abnormal or deficient functions of these enzymes. The availability of the rat genome sequence has opened the possibility to perform a global analysis of the complete protease repertoire or degradome of this model organism. The rat degradome consists of at least 626 proteases and homologs, which are distributed into five catalytic classes: 24 aspartic, 160 cysteine, 192 metallo, 221 serine, and 29 threonine proteases. Overall, this distribution is similar to that of the mouse degradome, but significatively more complex than that corresponding to the human degradome composed of 561 proteases and homologs. This increased complexity of the rat protease complement mainly derives from the expansion of several gene families including placental cathepsins, testases, kallikreins and hematopoietic serine proteases, involved in reproductive or immunological functions. These protease families have also evolved differently in the rat and mouse genomes and may contribute to explain some functional differences between these two closely related species. Likewise, genomic analysis of rat protease inhibitors has shown some differences with the mouse protease inhibitor complement and the marked expansion of families of cysteine and serine protease inhibitors in rat and mouse with respect to human. -

The Major Leucyl Aminopeptidase of Trypanosoma Cruzi (Laptc) Assembles Into a Homohexamer and Belongs to the M17 Family of Metal
Cadavid-Restrepo et al. BMC Biochemistry 2011, 12:46 http://www.biomedcentral.com/1471-2091/12/46 RESEARCH ARTICLE Open Access The major leucyl aminopeptidase of Trypanosoma cruzi (LAPTc) assembles into a homohexamer and belongs to the M17 family of metallopeptidases Gloria Cadavid-Restrepo1,2†, Thiago S Gastardelo1†, Eric Faudry3,4,5, Hugo de Almeida1, Izabela MD Bastos6, Raquel S Negreiros1, Meire M Lima1, Teresa C Assumpção1,7, Keyla C Almeida1, Michel Ragno3,4,5, Christine Ebel5,8,9 , Bergmann M Ribeiro1, Carlos R Felix1 and Jaime M Santana1* Abstract Background: Pathogens depend on peptidase activities to accomplish many physiological processes, including interaction with their hosts, highlighting parasitic peptidases as potential drug targets. In this study, a major leucyl aminopeptidolytic activity was identified in Trypanosoma cruzi, the aetiological agent of Chagas disease. Results: The enzyme was isolated from epimastigote forms of the parasite by a two-step chromatographic procedure and associated with a single 330-kDa homohexameric protein as determined by sedimentation velocity and light scattering experiments. Peptide mass fingerprinting identified the enzyme as the predicted T. cruzi aminopeptidase EAN97960. Molecular and enzymatic analysis indicated that this leucyl aminopeptidase of T. cruzi (LAPTc) belongs to the peptidase family M17 or leucyl aminopeptidase family. LAPTc has a strong dependence on neutral pH, is mesophilic and retains its oligomeric form up to 80°C. Conversely, its recombinant form is thermophilic and requires alkaline pH. Conclusions: LAPTc is a 330-kDa homohexameric metalloaminopeptidase expressed by all T. cruzi forms and mediates the major parasite leucyl aminopeptidolytic activity. Since biosynthetic pathways for essential amino acids, including leucine, are lacking in T. -

The Metalloaminopeptidases from Plasmodium Falciparum
Available online at www.sciencedirect.com Working in concert: the metalloaminopeptidases from Plasmodium falciparum Sheena McGowan Malaria remains the world’s most prevalent human parasitic Parasites of the genus Plasmodium are the causative agents disease. Because of the rapid spread of drug resistance in of malaria. While four Plasmodium species commonly infect parasites, there is an urgent need to identify diverse new drug humans, Plasmodium falciparum (Pf) causes the most targets. One group of proteases that are emerging as targets deaths [3–5]. To date, nine MAPs have been identified for novel antimalarials are the metalloaminopeptidases. These in Pf (3D7) [6]. Four of these are methionine aminopepti- enzymes catalyze the removal of the N-terminal amino acids dases. The other five enzymes comprise a prolyl imino- 2 from proteins and peptides. Given the restricted specificities of peptidase (or post-prolyl aminopeptidase), a prolyl each of these enzymes for different N-terminal amino acids, it is aminopeptidase (see footnote 2), a leucine aminopepti- thought that they act in concert to facilitate protein turnover. dase, an alanine aminopeptidase and an aspartyl amino- Here we review recent structure and functional data relating to peptidase. Given the restricted specificities of each of the development of the Plasmodium falciparum these enzymes for particular N-terminal amino acids, it metalloaminopeptidases as drug targets is thought that they act in concert to facilitate hemoglobin Addresses (Hb) catabolism [7,8], a process essential to the survival of Department of Biochemistry and Molecular Biology, Monash University, Pf. However, recent studies show that the PfMAPs may Clayton Campus, Melbourne, Victoria 3800, Australia have additional roles to that of Hb digestion, including important housekeeping functions and in conjunction with Corresponding author: McGowan, Sheena ([email protected]) the parasite proteosome [9 ,10]. -

Proline Residues in the Maturation and Degradation of Peptide Hormones and Neuropeptides
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Volume 234, number 2, 251-256 FEB 06071 July 1988 Review Letter Proline residues in the maturation and degradation of peptide hormones and neuropeptides Rolf Mentlein Anatomisches Institut der Christian-Albrechts Universitiit zu Kiel, OlshausenstraJe 40.2300 Kiel I, FRG Received 4 May 1988; revised version received 23 May 1988 The proteases involved in the maturation of regulatory peptides like those of broader specificity normally fail to cleave peptide bonds linked to the cyclic amino acid proline. This generates several mature peptides with N-terminal X-Pro- sequences. However, in certain non-mammalian tissues repetitive pre-sequences of this type are removed by specialized dipeptidyl (amino)peptidases during maturation. In mammals, proline-specific proteases are not involved in the biosyn- thesis of regulatory peptides, but due to their unique specificity they could play an important role in the degradation of them. Evidence exists that dipeptidyl (amino)peptidase IV at the cell surface of endothelial cells sequesters circulating peptide hormones which arc then susceptible to broader aminopeptidase attack. The cleavage of several neuropeptides by prolyl endopeptidase has been demonstrated in vitro, but its role in the brain is questionable since the precise localiza- tion of the protease is not clarified. Neuropeptide; Peptide hormone; Proteolytic processing; Degradation; Dipeptidyl (amino)peptidase; Prolyl endopeptidase 1. INTRODUCTION 2. DIRECTION OF THE PROTEOLYTIC MATURATION OF REGULATORY The unique cyclic and imino structure of the PEPTIDES BY PROLINE RESIDUES amino acid proline influences not only the confor- mation of peptide chains, but also restricts the at- Regulatory peptides are synthesized as part of tack of proteases. -

How Metal Cofactors Drive Dimer–Dodecamer Transition of the M42 Aminopeptidase Tmpep1050 of Thermotoga Maritima
This is a repository copy of How metal cofactors drive dimer–dodecamer transition of the M42 aminopeptidase TmPep1050 of Thermotoga maritima. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/159066/ Version: Accepted Version Article: Dutoit, R, Van Gompel, T, Brandt, N et al. (4 more authors) (2019) How metal cofactors drive dimer–dodecamer transition of the M42 aminopeptidase TmPep1050 of Thermotoga maritima. Journal of Biological Chemistry, 294 (47). pp. 17777-17789. ISSN 0021-9258 https://doi.org/10.1074/jbc.ra119.009281 © 2019 Dutoit et al. Published under exclusive license by The American Society for Biochemistry and Molecular Biology, Inc. This is an author produced version of a paper published in Journal of Biological Chemistry. Uploaded in accordance with the publisher's self-archiving policy. Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ -
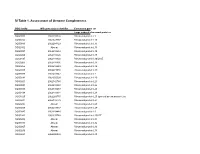
SI Table 1. Assessment of Genome Completeness
SI Table 1. Assessment of Genome Completeness COG family IMG gene object identifier Conserved gene set Large subunit ribosomal proteins COG0081 2062288324 Ribosomal protein L1 COG0244 2062347387 Ribosomal protein L10 COG0080 2062288323 Ribosomal protein L11 COG0102 Absent Ribosomal protein L13 COG0093 2062418832 Ribosomal protein L14 COG0200 2062418826 Ribosomal protein L15 COG0197 2062418838 Ribosomal protein L16/L10E COG0203 2062418836 Ribosomal protein L17 COG0256 2062418829 Ribosomal protein L18 COG0335 2062273558 Ribosomal protein L19 COG0090 2062418842 Ribosomal protein L2 COG0292 2062350539 Ribosomal protein L20 COG0261 2062142780 Ribosomal protein L21 COG0091 2062418840 Ribosomal protein L22 COG0089 2062138283 Ribosomal protein L23 COG0198 2062418834 Ribosomal protein L24 COG1825 2062269715 Ribosomal protein L25 (general stress protein Ctc) COG0211 2062142779 Ribosomal protein L27 COG0227 Absent Ribosomal protein L28 COG0255 2062418837 Ribosomal protein L29 COG0087 2062154483 Ribosomal protein L3 COG1841 2062335748 Ribosomal protein L30/L7E COG0254 Absent Ribosomal protein L31 COG0333 Absent Ribosomal protein L32 COG0267 Absent Ribosomal protein L33 COG0230 Absent Ribosomal protein L34 COG0291 2062350538 Ribosomal protein L35 COG0257 Absent Ribosomal protein L36 COG0088 2062138282 Ribosomal protein L4 COG0094 2062418833 Ribosomal protein L5 COG0097 2062418830 Ribosomal protein L6P/L9E COG0222 2062288326 Ribosomal protein L7/L12 COG0359 2062209880 Ribosomal protein L9 Small subunit ribosomal proteins COG0539 Absent Ribosomal protein