Functional Block Copolymers for Applications in Advanced Materials, Energy Storage, and Lithography Christopher George Hardy University of South Carolina
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Sustainable Triazine-Based Dehydro-Condensation Agents for Amide Synthesis
molecules Article Sustainable Triazine-Based Dehydro-Condensation Agents for Amide Synthesis Roberto Sole 1 , Vanessa Gatto 2 , Silvia Conca 1 , Noemi Bardella 1, Andrea Morandini 1 and Valentina Beghetto 1,2,* 1 Dipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari di Venezia, Via Torino 155, 30172 Venezia, Italy; [email protected] (R.S.); [email protected] (S.C.); [email protected] (N.B.); [email protected] (A.M.) 2 Crossing srl, Viale della Repubblica 193/b, 31100 Treviso, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-041-2348928 Abstract: Conventional methods employed today for the synthesis of amides often lack of economic and environmental sustainability. Triazine-derived quaternary ammonium salts, e.g., 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM(Cl)), emerged as promising dehydro-condensation agents for amide synthesis, although suffering of limited stability and high costs. In the present work, a simple protocol for the synthesis of amides mediated by 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) and a tert-amine has been described and data are compared to DMTMM(Cl) and other CDMT-derived quaternary ammonium salts (DMT-Ams(X), X: − − Cl or ClO4 ). Different tert-amines (Ams) were tested for the synthesis of various DMT-Ams(Cl), but only DMTMM(Cl) could be isolated and employed for dehydro-condensation reactions, while all CDMT/tert-amine systems tested were efficient as dehydro-condensation agents. Interestingly, in best reaction conditions, CDMT and 1,4-dimethylpiperazine gave N-phenethyl benzamide in 93% yield in 15 min, with up to half the amount of tert-amine consumption. -

The Reaction of Arenetricarbonylchromium Complexes with Alkyllithium Could Proceed Via a Variety of Pathways to Yield Any of Several Possible Products
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1975 The er action of arenetricarbonylchromium complexes with alkyllithium compounds Roger John Card Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Card, Roger John, "The er action of arenetricarbonylchromium complexes with alkyllithium compounds " (1975). Retrospective Theses and Dissertations. 5410. https://lib.dr.iastate.edu/rtd/5410 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This material was produced from a microfilm copy of the original document. While the most advance technological means to photograim and reproduce this document have been used, the quality is heavily dependent upon the quality of the original submitted. The following explanation of techniques is provided to help you understand markings or patterns which may appear on this reproduction. 1. The sign or "target" for pages apparency lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adiacent pages. This may have necessitated cutting tiiru an image and duplicating adjscsnt pages to insure you complete continuity. 2. When an image on the film is obliterated with a large round biack mark, it is an indication Aat the photographer suspected that the copy may have moved during exposure and thus cause a blurred image. -

EI-ICHI NEGISHI Herbert C
MAGICAL POWER OF TRANSITION METALS: PAST, PRESENT, AND FUTURE Nobel Lecture, December 8, 2010 by EI-ICHI NEGISHI Herbert C. Brown Laboratories of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907-2084, U.S.A. Not long ago, the primary goal of the synthesis of complex natural products and related compounds of biological and medicinal interest was to be able to synthesize them, preferably before anyone else. While this still remains a very important goal, a number of today’s top-notch synthetic chemists must feel and even think that, given ample resources and time, they are capable of synthesizing virtually all natural products and many analogues thereof. Accepting this notion, what would then be the major goals of organic synthesis in the twenty-first century? One thing appears to be unmistakably certain. Namely, we will always need, perhaps increasingly so with time, the uniquely creative field of synthetic organic and organometallic chemistry to prepare both new and existing organic compounds for the benefit and well-being of mankind. It then seems reasonably clear that, in addition to the question of what compounds to synthesize, that of how best to synthesize them will become increasingly important. As some may have said, the primary goal would then shift from aiming to be the first to synthesize a given compound to seeking its ultimately satisfactory or “last synthesis”. If one carefully goes over various aspects of organic synthetic methodology, one would soon note how primitive and limited it had been until rather recently, or perhaps even today. For the sake of argument, we may propose here that the ultimate goal of organic synthesis is “to be able to synthesize any desired and fundamentally synthesizable organic compounds (a) in high yields, (b) efficiently (in as few steps as possible, for example), (c) selectively, preferably all in t98–99% selectivity, (d) economically, and (e) safely, abbreviated hereafter as the y(es)2 manner.” with or without catalyst R1M + R2X R1R2 + MX R1, R2: carbon groups. -

Catalytic Systems Based on Cp2zrx2 (X = Cl, H), Organoaluminum
catalysts Article Catalytic Systems Based on Cp2ZrX2 (X = Cl, H), Organoaluminum Compounds and Perfluorophenylboranes: Role of Zr,Zr- and Zr,Al-Hydride Intermediates in Alkene Dimerization and Oligomerization Lyudmila V. Parfenova 1,* , Pavel V. Kovyazin 1, Almira Kh. Bikmeeva 1 and Eldar R. Palatov 2 1 Institute of Petrochemistry and Catalysis of Russian Academy of Sciences, Prospekt Oktyabrya, 141, 450075 Ufa, Russia; [email protected] (P.V.K.); [email protected] (A.K.B.) 2 Bashkir State University, st. Zaki Validi, 32, 450076 Ufa, Russia; [email protected] * Correspondence: [email protected]; Tel.: +7-347-284-3527 i i Abstract: The activity and chemoselectivity of the Cp2ZrCl2-XAlBu 2 (X = H, Bu ) and [Cp2ZrH2]2- ClAlEt2 catalytic systems activated by (Ph3C)[B(C6F5)4] or B(C6F5)3 were studied in reactions with 1-hexene. The activation of the systems by B(C6F5)3 resulted in the selective formation of head- to-tail alkene dimers in up to 93% yields. NMR studies of the reactions of Zr complexes with organoaluminum compounds (OACs) and boron activators showed the formation of Zr,Zr- and Zr,Al-hydride intermediates, for which diffusion coefficients, hydrodynamic radii, and volumes were estimated using the diffusion ordered spectroscopy DOSY. Bis-zirconium hydride clusters of type x[Cp ZrH ·Cp ZrHCl·ClAlR ]·yRnAl(C F ) − were found to be the key intermediates of alkene 2 2 2 2 6 5 3 n dimerization, whereas cationic Zr,Al-hydrides led to the formation of oligomers. Citation: Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R. -

Organometrallic Chemistry
CHE 425: ORGANOMETALLIC CHEMISTRY SOURCE: OPEN ACCESS FROM INTERNET; Striver and Atkins Inorganic Chemistry Lecturer: Prof. O. G. Adeyemi ORGANOMETALLIC CHEMISTRY Definitions: Organometallic compounds are compounds that possess one or more metal-carbon bond. The bond must be “ionic or covalent, localized or delocalized between one or more carbon atoms of an organic group or molecule and a transition, lanthanide, actinide, or main group metal atom.” Organometallic chemistry is often described as a bridge between organic and inorganic chemistry. Organometallic compounds are very important in the chemical industry, as a number of them are used as industrial catalysts and as a route to synthesizing drugs that would not have been possible using purely organic synthetic routes. Coordinative unsaturation is a term used to describe a complex that has one or more open coordination sites where another ligand can be accommodated. Coordinative unsaturation is a very important concept in organotrasition metal chemistry. Hapticity of a ligand is the number of atoms that are directly bonded to the metal centre. Hapticity is denoted with a Greek letter η (eta) and the number of bonds a ligand has with a metal centre is indicated as a superscript, thus η1, η2, η3, ηn for hapticity 1, 2, 3, and n respectively. Bridging ligands are normally preceded by μ, with a subscript to indicate the number of metal centres it bridges, e.g. μ2–CO for a CO that bridges two metal centres. Ambidentate ligands are polydentate ligands that can coordinate to the metal centre through one or more atoms. – – – For example CN can coordinate via C or N; SCN via S or N; NO2 via N or N. -

Cyclopentadienyl Compounds of the First Row Transition Metals And
University of Vermont ScholarWorks @ UVM Graduate College Dissertations and Theses Dissertations and Theses 2017 Cyclopentadienyl Compounds of the First Row Transition Metals and Early Actinides: Novel Main-Group Bond Forming Catalysis and New Metallacycles Justin Kane Pagano University of Vermont Follow this and additional works at: https://scholarworks.uvm.edu/graddis Part of the Chemistry Commons Recommended Citation Pagano, Justin Kane, "Cyclopentadienyl Compounds of the First Row Transition Metals and Early Actinides: Novel Main-Group Bond Forming Catalysis and New Metallacycles" (2017). Graduate College Dissertations and Theses. 700. https://scholarworks.uvm.edu/graddis/700 This Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks @ UVM. It has been accepted for inclusion in Graduate College Dissertations and Theses by an authorized administrator of ScholarWorks @ UVM. For more information, please contact [email protected]. CYCLOPENTADIENYL COMPOUNDS OF THE FIRST ROW TRANSITION METALS AND EARLY ACTINIDES: NOVEL MAIN-GROUP BOND FORMING CATALYSIS AND NEW METALLACYCLES A Dissertation Presented by Justin Kane Pagano to The Faculty of the Graduate College of The University of Vermont In Partial Fulfilment of the Requirements For the Degree of Doctor of Philosophy Specializing in Chemistry May, 2017 Defense Date: November 29, 2016 Dissertation Examination Committee: Rory Waterman, Ph. D., Advisor John M. Hughes, Ph. D., Chairperson Matthias Brewer, Ph. D. Jaqueline L. Kiplinger, Ph. D. Matthew D. Liptak, Ph. D. Cynthia J. Forehand, Ph. D., Dean of the Graduate College ABSTRACT Cyclopentadienyl first row transition-metal compounds have been well studied 5 since the 1950’s, with the nearly ubiquitous CpFe(CO)2Me (FpMe) (Cp = η -C5H5) being one of the first organometallics to be fully characterized. -

Supporting Information Degradation
Electronic Supplementary Material (ESI) for Catalysis Science & Technology. This journal is © The Royal Society of Chemistry 2021 Supporting Information Degradation Pathways of a highly active iron(III) tetra-NHC Epoxidation Catalyst Florian Dyckhoff, Jonas F. Schlagintweit, Marco A. Bernd, Christian H. G. Jakob, Tim P. Schlachta, Benjamin J. Hofmann, Robert M. Reich and Fritz E. Kühn* Molecular Catalysis, Catalysis Research Center and Department of Chemistry, Technische Universität München, Lichtenbergstrasse 4, D-85748 Garching bei München, Germany. [*] Corresponding author: Prof. Dr. Fritz E. Kühn, E-Mail: [email protected] Table of Contents 1. Effects of bases on 2, 2-tBuCN and 2-PhCN – Catalysis and UV/vis Spectroscopy 2 2. 1H-NMR Spectroscopic Studies 5 3. Synthesis and Characterization of compound 2-d8 7 4. ESI-MS Decomposition Studies 27 5. DFT Calculations 31 6. References 34 1 1. Effects of bases on 2, 2-tBuCN and 2-PhCN – Catalysis and UV/vis Spectroscopy All batch and time-dependent reactions were conducted in a cryostat (Julabo FP-50) with a total reaction volume of 2.0 mL. The catalyst (0.05 mol%, 0.067 µmol) was added from a preformed stock solution (4.0 mg/mL in the respective nitrile, i.e. acetonitrile, tert-butylnitrile or benzonitrile) according to the appropriate stoichiometry to a solution of cis-cyclooctene (100 mol%, 134.5 µmol), the respective additive (0.5 mol%, 0.67 µmol) and H2O2 (150 mol%, 202 µmol, 50% solution in H2O) in the appropriate solvent. The reaction was initiated upon addition of H2O2. The reaction was aborted by adding electrolytically precipitated activated MnO2 as a H2O2 decomposition agent. -

Synthesis and Reactivity of Cyclopentadienyl Based Organometallic Compounds and Their Electrochemical and Biological Properties
Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Sasmita Mishra Department of Chemistry National Institute of Technology Rourkela Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties Dissertation submitted to the National Institute of Technology Rourkela In partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry by Sasmita Mishra (Roll Number: 511CY604) Under the supervision of Prof. Saurav Chatterjee February, 2017 Department of Chemistry National Institute of Technology Rourkela Department of Chemistry National Institute of Technology Rourkela Certificate of Examination Roll Number: 511CY604 Name: Sasmita Mishra Title of Dissertation: ''Synthesis and reactivity of cyclopentadienyl based organometallic compounds and their electrochemical and biological properties We the below signed, after checking the dissertation mentioned above and the official record book(s) of the student, hereby state our approval of the dissertation submitted in partial fulfillment of the requirements of the degree of Doctor of Philosophy in Chemistry at National Institute of Technology Rourkela. We are satisfied with the volume, quality, correctness, and originality of the work. --------------------------- Prof. Saurav Chatterjee Principal Supervisor --------------------------- --------------------------- Prof. A. Sahoo. Prof. G. Hota Member (DSC) Member (DSC) --------------------------- -

Chapter 2 Improvement of Heat Resistance of Poly(Methyl Methacrylate) by Addition of Lithium Salts
JAIST Repository https://dspace.jaist.ac.jp/ リチウム塩の添加がポリメタクリル酸メチルの特性に Title 及ぼす影響 Author(s) 伊藤, 麻絵 Citation Issue Date 2019-03 Type Thesis or Dissertation Text version ETD URL http://hdl.handle.net/10119/15795 Rights Description Supervisor:山口 政之, 先端科学技術研究科, 博士 Japan Advanced Institute of Science and Technology Effect of addition of lithium salts on properties of poly(methyl methacrylate) Asae Ito Japan Advanced Institute of Science and Technology Doctoral Dissertation Effect of addition of lithium salts on properties of poly(methyl methacrylate) Asae Ito Supervisor: Prof. Dr. Masayuki Yamaguchi Graduate School of Advanced Science and Technology Japan Advanced Institute of Science and Technology Materials Science March 2019 Referee-in-chief: Professor Masayuki Yamaguchi Japan Advanced Institute of Science and Technology Referees: Professor Masayuki Yamaguchi Japan Advanced Institute of Science and Technology Professor Tatsuo Kaneko Japan Advanced Institute of Science and Technology Associate Professor Toshiaki Taniike Japan Advanced Institute of Science and Technology Associate Professor Ken-ichi Shinohara Japan Advanced Institute of Science and Technology Professor Akihiro Nishioka Yamagata University Effect of addition of lithium salts on properties of poly(methyl methacrylate) Yamaguchi Laboratory Asae Ito (s1620002) Heat resistance and optical properties of amorphous polymers are important for engineering applications such as automobile parts, electrical devices, and displays. In general, a single plastic material often possesses poor physical properties for engineering application, so that the improvement of properties has been tried in decades. In particular, one of the promising processes to modify a polymer is mixing with low-molecular-weight compounds. Recently, it was found that the addition of a specific lithium salt enhances glass transition temperature (Tg) of poly(methyl methacrylate) (PMMA), which is a typical amorphous polymer. -

US20100160506A1.Pdf
US 2010.0160506A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0160506 A1 Wu et al. (43) Pub. Date: Jun. 24, 2010 (54) PRODUCTION OFSYNTHETIC Publication Classification HYDROCARBON FLUIDS, PLASTICIZERS (51) Int. Cl. AND SYNTHETICLUBRICANT BASE CSK 5/55 (2006.01) STOCKS FROM RENEWABLE FEEDSTOCKS CSK 5/109 (2006.01) C07D 30/03 (2006.01) (76) Inventors: Margaret May-Som Wu, Skillman, C07D 31 7/24 (2006.01) NJ (US); Karla Schall Colle, C07C I/207 (2006.01) Houston, TX (US); Ramzi Yanni (52) U.S. Cl. ......... 524/114; 524/280; 549/523: 549/229; Saleh, Baton Rouge, LA (US); 585/327 Allen D. Godwin, Seabrook, TX (57) ABSTRACT (US); John Edmond Randolph This disclosure is directed to an integrated method for making Stanat, Houston, TX (US) synthetic hydrocarbon fluids, plasticizers and polar synthetic lubricant base stocks from a renewable feedstock. More par Correspondence Address: ticularly, the disclosure is directed to a metathesis reaction of ExxonMobil Research & Engineering Company natural oil or its derivative ester and ethylene in the presence P.O. Box 900, 1545 Route 22 East of an effective amount of a metathesis catalyst to form linear Annandale, NJ 08801-0900 (US) alpha-olefins, internal olefins and reduced chain length trig lycerides. The linear alpha-olefins and/or internal olefins are polymerized to produce synthetic hydrocarbon fluids in the (21) Appl. No.: 12/633,742 presence of a suitable catalyst. The reduced chain length triglycerides are converted into polar synthetic lubricant base (22) Filed: Dec. 17, 2009 stocks or plasticizers by hydrogenation, isomerization, fol lowed by hydrogenations, or by hydroisomerization pro cesses. -

ACS Guide to Scholarly Com
1.3 Communicating Safety Information Samuella Sigmann Visit the full ACS Guide to Scholarly Communication 1.3.1 Introduction In 2011, the U.S. Chemical Safety Board (CSB) issued its first investigation of a chemical accident at an academic research institution. The incident involved the sudden detonation of a nickel hydrazine perchlorate (NHP) derivative that resulted in severe and permanent injury to a fifth-year chemistry graduate student at Texas Tech University (TTU). Student researchers were involved in a multi-institutional research project to characterize new potentially energetic materials. They decided to scale up their synthesis by two orders of magnitude to provide enough sample so that all testing could be performed on one batch but neglected to assess safety ramifications. The final CSB report for the TTU incident found in both this case and prior incidents in the same research area that (1): . No formal hazard evaluation and risk assessment had been completed to char-acterize the potential danger of the research activity and to plan for the worst-case scenario. The student sought peer advice. No policy was in place at the laboratory, department, or university level to prompt a student to seek PI advice or evaluation of experimental activities. The investigation clearly indicates that the students did not have the information they needed to prepare themselves for the increased risk posed by their decision to scale up. The CSB primarily investigates and reports on large scale incidents in the chemical and related industries where hazard evaluation and risk assessment are well established to facilitate improved safety management across the industry. -
Main Group Metallocenes
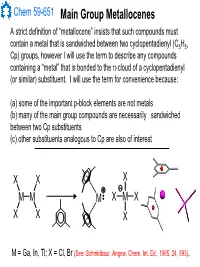
Chem 59-651 Main Group Metallocenes A strict definition of “metallocene” insists that such compounds must contain a metal that is sandwiched between two cyclopentadienyl (C5H5, Cp) groups, however I will use the term to describe any compounds containing a “metal” that is bonded to the π-cloud of a cyclopentadienyl (or similar) substituent. I will use the term for convenience because: (a) some of the important p-block elements are not metals (b) many of the main group compounds are necessarily sandwiched between two Cp substituents (c) other substituents analogous to Cp are also of interest XX X MM M XMX X X X M = Ga, In, Tl; X = Cl, Br (See: Schmidbaur, Angew. Chem. Int. Ed., 1985, 24, 893). Chem 59-651 Cyclopentadienyl Ligand Basics The structural features and bonding interactions between cyclopentadienyl groups and metal atoms shows considerable variation and is described using a few different conventions (See Jutzi and Burford, Chem. Rev., 1999, 99, 969 and references therein). In most main group metallocenes, even those that are σ-bonded in the solid state, the rings rotate rapidly through a series of 1,2-shifts. A Cp group can be considered to be p-bonded to an element if the Hapticity: element sits inside the cylinder defined by the 5 carbon atoms of M MM the Cp ligand. The center of mass of the 5 C atoms is known as the centroid. η1-Cp η2-Cp η3-Cp M M M A σ-bonded Cp can be identified η4-Cp η5-Cp ≡η5-Cp by the angle at the ipso carbon M and bond lengths should indicate single bonds to the ipso carbon and a localized diene structure for σ the α and β carbon fragment.