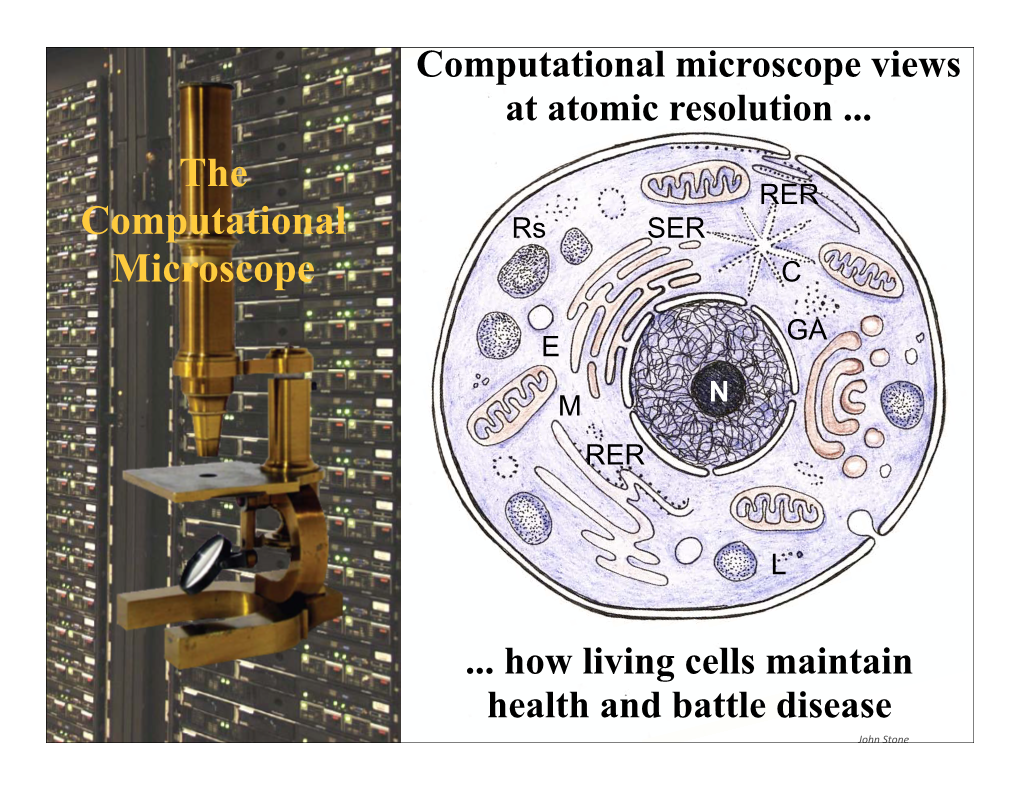
The Computational Microscope
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Real-Time Pymol Visualization for Rosetta and Pyrosetta
Real-Time PyMOL Visualization for Rosetta and PyRosetta Evan H. Baugh1, Sergey Lyskov1, Brian D. Weitzner1, Jeffrey J. Gray1,2* 1 Department of Chemical and Biomolecular Engineering, The Johns Hopkins University, Baltimore, Maryland, United States of America, 2 Program in Molecular Biophysics, The Johns Hopkins University, Baltimore, Maryland, United States of America Abstract Computational structure prediction and design of proteins and protein-protein complexes have long been inaccessible to those not directly involved in the field. A key missing component has been the ability to visualize the progress of calculations to better understand them. Rosetta is one simulation suite that would benefit from a robust real-time visualization solution. Several tools exist for the sole purpose of visualizing biomolecules; one of the most popular tools, PyMOL (Schro¨dinger), is a powerful, highly extensible, user friendly, and attractive package. Integrating Rosetta and PyMOL directly has many technical and logistical obstacles inhibiting usage. To circumvent these issues, we developed a novel solution based on transmitting biomolecular structure and energy information via UDP sockets. Rosetta and PyMOL run as separate processes, thereby avoiding many technical obstacles while visualizing information on-demand in real-time. When Rosetta detects changes in the structure of a protein, new coordinates are sent over a UDP network socket to a PyMOL instance running a UDP socket listener. PyMOL then interprets and displays the molecule. This implementation also allows remote execution of Rosetta. When combined with PyRosetta, this visualization solution provides an interactive environment for protein structure prediction and design. Citation: Baugh EH, Lyskov S, Weitzner BD, Gray JJ (2011) Real-Time PyMOL Visualization for Rosetta and PyRosetta. -

An Explicit-Solvent Conformation Search Method Using Open Software
An explicit-solvent conformation search method using open software Kari Gaalswyk and Christopher N. Rowley Department of Chemistry, Memorial University of Newfoundland, St. John’s, Newfoundland and Labrador, Canada ABSTRACT Computer modeling is a popular tool to identify the most-probable conformers of a molecule. Although the solvent can have a large effect on the stability of a conformation, many popular conformational search methods are only capable of describing molecules in the gas phase or with an implicit solvent model. We have developed a work-flow for performing a conformation search on explicitly-solvated molecules using open source software. This method uses replica exchange molecular dynamics (REMD) to sample the conformational states of the molecule efficiently. Cluster analysis is used to identify the most probable conformations from the simulated trajectory. This work-flow was tested on drug molecules a-amanitin and cabergoline to illustrate its capabilities and effectiveness. The preferred conformations of these molecules in gas phase, implicit solvent, and explicit solvent are significantly different. Subjects Biophysics, Pharmacology, Computational Science Keywords Conformation search, Explicit solvent, Cluster analysis, Replica exchange molecular dynamics INTRODUCTION Many molecules can exist in multiple conformational isomers. Conformational isomers have the same chemical bonds, but differ in their 3D geometry because they hold different torsional angles (Crippen & Havel, 1988). The conformation of a molecule can affect Submitted 5 April 2016 chemical reactivity, molecular binding, and biological activity (Struthers, Rivier & Accepted 6May2016 Published 31 May 2016 Hagler, 1985; Copeland, 2011). Conformations differ in stability because they experience different steric, electrostatic, and solute-solvent interactions. The probability, p,ofa Corresponding author Christopher N. -

Python Tools in Computational Chemistry (And Biology)
Python Tools in Computational Chemistry (and Biology) Andrew Dalke Dalke Scientific, AB Göteborg, Sweden EuroSciPy, 26-27 July, 2008 “Why does ‘import numpy’ take 0.4 seconds? Does it need to import 228 libraries?” - My first Numpy-discussion post (paraphrased) Your use case isn't so typical and so suffers on the import time end of the balance. - Response from Robert Kern (Others did complain. Import time down to 0.28s.) 52,000 structures PDB doubles every 2½ years HEADER PHOTORECEPTOR 23-MAY-90 1BRD 1BRD 2 COMPND BACTERIORHODOPSIN 1BRD 3 SOURCE (HALOBACTERIUM $HALOBIUM) 1BRD 4 EXPDTA ELECTRON DIFFRACTION 1BRD 5 AUTHOR R.HENDERSON,J.M.BALDWIN,T.A.CESKA,F.ZEMLIN,E.BECKMANN, 1BRD 6 AUTHOR 2 K.H.DOWNING 1BRD 7 REVDAT 3 15-JAN-93 1BRDB 1 SEQRES 1BRDB 1 REVDAT 2 15-JUL-91 1BRDA 1 REMARK 1BRDA 1 .. ATOM 54 N PRO 8 20.397 -15.569 -13.739 1.00 20.00 1BRD 136 ATOM 55 CA PRO 8 21.592 -15.444 -12.900 1.00 20.00 1BRD 137 ATOM 56 C PRO 8 21.359 -15.206 -11.424 1.00 20.00 1BRD 138 ATOM 57 O PRO 8 21.904 -15.930 -10.563 1.00 20.00 1BRD 139 ATOM 58 CB PRO 8 22.367 -14.319 -13.591 1.00 20.00 1BRD 140 ATOM 59 CG PRO 8 22.089 -14.564 -15.053 1.00 20.00 1BRD 141 ATOM 60 CD PRO 8 20.647 -15.054 -15.103 1.00 20.00 1BRD 142 ATOM 61 N GLU 9 20.562 -14.211 -11.095 1.00 20.00 1BRD 143 ATOM 62 CA GLU 9 20.192 -13.808 -9.737 1.00 20.00 1BRD 144 ATOM 63 C GLU 9 19.567 -14.935 -8.932 1.00 20.00 1BRD 145 ATOM 64 O GLU 9 19.815 -15.104 -7.724 1.00 20.00 1BRD 146 ATOM 65 CB GLU 9 19.248 -12.591 -9.820 1.00 99.00 1 1BRD 147 ATOM 66 CG GLU 9 19.902 -11.351 -10.387 1.00 99.00 1 1BRD 148 ATOM 67 CD GLU 9 19.243 -10.169 -10.980 1.00 99.00 1 1BRD 149 ATOM 68 OE1 GLU 9 18.323 -10.191 -11.782 1.00 99.00 1 1BRD 150 ATOM 69 OE2 GLU 9 19.760 -9.089 -10.597 1.00 99.00 1 1BRD 151 ATOM 70 N TRP 10 18.764 -15.737 -9.597 1.00 20.00 1BRD 152 ATOM 71 CA TRP 10 18.034 -16.884 -9.090 1.00 20.00 1BRD 153 ATOM 72 C TRP 10 18.843 -17.908 -8.318 1.00 20.00 1BRD 154 ATOM 73 O TRP 10 18.376 -18.310 -7.230 1.00 20.00 1BRD 155 . -

Computational Chemistry for Chemistry Educators Shawn C
Volume 1, Issue 1 Journal Of Computational Science Education Computational Chemistry for Chemistry Educators Shawn C. Sendlinger Clyde R. Metz North Carolina Central University College of Charleston Department of Chemistry Department of Chemistry and Biochemistry 1801 Fayetteville Street, 66 George Street, Durham, NC 27707 Charleston, SC 29424 919-530-6297 843-953-8097 [email protected] [email protected] ABSTRACT 1. INTRODUCTION In this paper we describe an ongoing project where the goal is to The majority of today’s students are technologically savvy and are develop competence and confidence among chemistry faculty so often more comfortable using computers than the faculty who they are able to utilize computational chemistry as an effective teach them. In order to harness the student’s interest in teaching tool. Advances in hardware and software have made technology and begin to use it as an educational tool, most faculty research-grade tools readily available to the academic community. members require some level of instruction and training. Because Training is required so that faculty can take full advantage of this chemistry research increasingly utilizes computation as an technology, begin to transform the educational landscape, and important tool, our approach to chemistry education should reflect attract more students to the study of science. this. The ability of computer technology to visualize and manipulate objects on an atomic scale can be a powerful tool to increase both student interest in chemistry as well as their level of Categories and Subject Descriptors understanding. Computational Chemistry for Chemistry Educators (CCCE) is a project that seeks to provide faculty the J.2 [Physical Sciences and Engineering]: Chemistry necessary knowledge, experience, and technology access so that they can begin to design and incorporate computational approaches in the courses they teach. -

Preparing and Analyzing Large Molecular Simulations With
Preparing and Analyzing Large Molecular Simulations with VMD John Stone, Senior Research Programmer NIH Center for Macromolecular Modeling and Bioinformatics, University of Illinois at Urbana-Champaign VMD Tutorials Home Page • http://www.ks.uiuc.edu/Training/Tutorials/ – Main VMD tutorial – VMD images and movies tutorial – QwikMD simulation preparation and analysis plugin – Structure check – VMD quantum chemistry visualization tutorial – Visualization and analysis of CPMD data with VMD – Parameterizing small molecules using ffTK Overview • Introduction • Data model • Visualization • Scripting and analytical features • Trajectory analysis and visualization • High fidelity ray tracing • Plugins and user-extensibility • Large system analysis and visualization VMD – “Visual Molecular Dynamics” • 100,000 active users worldwide • Visualization and analysis of: – Molecular dynamics simulations – Lattice cell simulations – Quantum chemistry calculations – Cryo-EM densities, volumetric data • User extensible scripting and plugins • http://www.ks.uiuc.edu/Research/vmd/ Cell-Scale Modeling MD Simulation VMD – “Visual Molecular Dynamics” • Unique capabilities: • Visualization and analysis of: – Trajectories are fundamental to VMD – Molecular dynamics simulations – Support for very large systems, – “Particle” systems and whole cells now reaching billions of particles – Cryo-EM densities, volumetric data – Extensive GPU acceleration – Quantum chemistry calculations – Parallel analysis/visualization with MPI – Sequence information MD Simulations Cell-Scale -

Kepler Gpus and NVIDIA's Life and Material Science
LIFE AND MATERIAL SCIENCES Mark Berger; [email protected] Founded 1993 Invented GPU 1999 – Computer Graphics Visual Computing, Supercomputing, Cloud & Mobile Computing NVIDIA - Core Technologies and Brands GPU Mobile Cloud ® ® GeForce Tegra GRID Quadro® , Tesla® Accelerated Computing Multi-core plus Many-cores GPU Accelerator CPU Optimized for Many Optimized for Parallel Tasks Serial Tasks 3-10X+ Comp Thruput 7X Memory Bandwidth 5x Energy Efficiency How GPU Acceleration Works Application Code Compute-Intensive Functions Rest of Sequential 5% of Code CPU Code GPU CPU + GPUs : Two Year Heart Beat 32 Volta Stacked DRAM 16 Maxwell Unified Virtual Memory 8 Kepler Dynamic Parallelism 4 Fermi 2 FP64 DP GFLOPS GFLOPS per DP Watt 1 Tesla 0.5 CUDA 2008 2010 2012 2014 Kepler Features Make GPU Coding Easier Hyper-Q Dynamic Parallelism Speedup Legacy MPI Apps Less Back-Forth, Simpler Code FERMI 1 Work Queue CPU Fermi GPU CPU Kepler GPU KEPLER 32 Concurrent Work Queues Developer Momentum Continues to Grow 100M 430M CUDA –Capable GPUs CUDA-Capable GPUs 150K 1.6M CUDA Downloads CUDA Downloads 1 50 Supercomputer Supercomputers 60 640 University Courses University Courses 4,000 37,000 Academic Papers Academic Papers 2008 2013 Explosive Growth of GPU Accelerated Apps # of Apps Top Scientific Apps 200 61% Increase Molecular AMBER LAMMPS CHARMM NAMD Dynamics GROMACS DL_POLY 150 Quantum QMCPACK Gaussian 40% Increase Quantum Espresso NWChem Chemistry GAMESS-US VASP CAM-SE 100 Climate & COSMO NIM GEOS-5 Weather WRF Chroma GTS 50 Physics Denovo ENZO GTC MILC ANSYS Mechanical ANSYS Fluent 0 CAE MSC Nastran OpenFOAM 2010 2011 2012 SIMULIA Abaqus LS-DYNA Accelerated, In Development NVIDIA GPU Life Science Focus Molecular Dynamics: All codes are available AMBER, CHARMM, DESMOND, DL_POLY, GROMACS, LAMMPS, NAMD Great multi-GPU performance GPU codes: ACEMD, HOOMD-Blue Focus: scaling to large numbers of GPUs Quantum Chemistry: key codes ported or optimizing Active GPU acceleration projects: VASP, NWChem, Gaussian, GAMESS, ABINIT, Quantum Espresso, BigDFT, CP2K, GPAW, etc. -
![Trends in Atomistic Simulation Software Usage [1.3]](https://docslib.b-cdn.net/cover/7978/trends-in-atomistic-simulation-software-usage-1-3-1207978.webp)
Trends in Atomistic Simulation Software Usage [1.3]
A LiveCoMS Perpetual Review Trends in atomistic simulation software usage [1.3] Leopold Talirz1,2,3*, Luca M. Ghiringhelli4, Berend Smit1,3 1Laboratory of Molecular Simulation (LSMO), Institut des Sciences et Ingenierie Chimiques, Valais, École Polytechnique Fédérale de Lausanne, CH-1951 Sion, Switzerland; 2Theory and Simulation of Materials (THEOS), Faculté des Sciences et Techniques de l’Ingénieur, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland; 3National Centre for Computational Design and Discovery of Novel Materials (MARVEL), École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland; 4The NOMAD Laboratory at the Fritz Haber Institute of the Max Planck Society and Humboldt University, Berlin, Germany This LiveCoMS document is Abstract Driven by the unprecedented computational power available to scientific research, the maintained online on GitHub at https: use of computers in solid-state physics, chemistry and materials science has been on a continuous //github.com/ltalirz/ rise. This review focuses on the software used for the simulation of matter at the atomic scale. We livecoms-atomistic-software; provide a comprehensive overview of major codes in the field, and analyze how citations to these to provide feedback, suggestions, or help codes in the academic literature have evolved since 2010. An interactive version of the underlying improve it, please visit the data set is available at https://atomistic.software. GitHub repository and participate via the issue tracker. This version dated August *For correspondence: 30, 2021 [email protected] (LT) 1 Introduction Gaussian [2], were already released in the 1970s, followed Scientists today have unprecedented access to computa- by force-field codes, such as GROMOS [3], and periodic tional power. -

Methods for the Refinement of Protein Structure 3D Models
International Journal of Molecular Sciences Review Methods for the Refinement of Protein Structure 3D Models Recep Adiyaman and Liam James McGuffin * School of Biological Sciences, University of Reading, Reading RG6 6AS, UK; [email protected] * Correspondence: l.j.mcguffi[email protected]; Tel.: +44-0-118-378-6332 Received: 2 April 2019; Accepted: 7 May 2019; Published: 1 May 2019 Abstract: The refinement of predicted 3D protein models is crucial in bringing them closer towards experimental accuracy for further computational studies. Refinement approaches can be divided into two main stages: The sampling and scoring stages. Sampling strategies, such as the popular Molecular Dynamics (MD)-based protocols, aim to generate improved 3D models. However, generating 3D models that are closer to the native structure than the initial model remains challenging, as structural deviations from the native basin can be encountered due to force-field inaccuracies. Therefore, different restraint strategies have been applied in order to avoid deviations away from the native structure. For example, the accurate prediction of local errors and/or contacts in the initial models can be used to guide restraints. MD-based protocols, using physics-based force fields and smart restraints, have made significant progress towards a more consistent refinement of 3D models. The scoring stage, including energy functions and Model Quality Assessment Programs (MQAPs) are also used to discriminate near-native conformations from non-native conformations. Nevertheless, there are often very small differences among generated 3D models in refinement pipelines, which makes model discrimination and selection problematic. For this reason, the identification of the most native-like conformations remains a major challenge. -

A Coarse-Grained Molecular Dynamics Simulation Using NAMD Package to Reveal Aggregation Profile of Phospholipids Self-Assembly in Water
Hindawi Publishing Corporation Journal of Chemistry Volume 2014, Article ID 273084, 6 pages http://dx.doi.org/10.1155/2014/273084 Research Article A Coarse-Grained Molecular Dynamics Simulation Using NAMD Package to Reveal Aggregation Profile of Phospholipids Self-Assembly in Water Dwi Hudiyanti,1 Muhammad Radifar,2 Tri Joko Raharjo,3 Narsito Narsito,3 and Sri Noegrohati4 1 Department of Chemistry, Diponegoro University, Jl. Prof. Sudharto, SH., Semarang 50257, Indonesia 2 Graduate School, Gadjah Mada University, Sekip Utara, Yogyakarta 55281, Indonesia 3 Department of Chemistry, FMIPA, Gadjah Mada University, Sekip Utara, Yogyakarta 55281, Indonesia 4 Faculty of Pharmacy, Gadjah Mada University, Sekip Utara, Yogyakarta 55281, Indonesia Correspondence should be addressed to Dwi Hudiyanti; dwi [email protected] Received 21 May 2014; Revised 11 July 2014; Accepted 14 July 2014; Published 4 August 2014 Academic Editor: Hugo Verli Copyright © 2014 Dwi Hudiyanti et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The energy profile of self-assembly process of DLPE, DLPS, DOPE, DOPS, DLiPE, and DLiPS in water was investigated by a coarse-grained molecular dynamics simulation using NAMD package. The self-assembly process was initiated from random configurations. The simulation was carried out for 160 ns. This study presented proof that there were three major self-assembled arrangements which became visible for a certain duration when the simulation took place, that is, liposome, deformed liposome, and planar bilayer. The energy profile that shows plateau at the time of these structures emerge confirmed their stability therein. -

Statistics of Software Package Usage in Supercomputer Complexes
Statistics of Software Package Usage in Supercomputer Complexes Pavel Shvets, Vadim Voevodin, and Sergey Zhumatiy Research Computing Center of Lomonosov Moscow State University, Moscow, Russia [email protected] Abstract. Any modern high-performance computing system is a very sophisticated complex that contains a variety of hardware and software components. Performance of such systems can be high, but it reaches its maximum only if all components work consistently and parallel applica- tions efficiently use provided computational resources. The steady trend of all recent years concerning large supercomputing centers { constant growth of the share of supercomputer applications that use off-the-shelf application packages. From this point of view, supercomputer software infrastructure starts to play a significant role in the efficient supercom- puter management as a whole. In this paper we will show why the analysis of software package efficiency should be taken seriously, what is worth paying attention to, and how this type of analysis can be conducted within the supercomputer complex. Keywords: parallel computing · high-performance computing · super- computer · efficiency · efficiency analysis · software package · job flow · monitoring data 1 Introduction Low efficiency of supercomputer system functioning has been an actual problem in high-performance computing field for many years. Over the years, many pos- sible approaches and methods have been developed for analysis and optimization of both individual supercomputer applications and entire supercomputer func- tioning as a whole. Due to increasing large-scale use of this area, i.e. growing number of users of supercomputer systems, as well as the progress in the develop- ment of HPC packages, off-the-shelf software packages are more and more often used for solving different scientific problems. -

Chemistry 6V39
Chemistry 6V91, 4V01 “Topics in Organic Chemistry: Computational Modeling” Spring 2006 – MW 5:30-6:45 PM, BE3.102 Instructors: Richard A. Caldwell, BE3.514, 972-883-2906, [email protected] Steven O. Nielsen, BE2.324, 972-883-6531, [email protected] Web site: http:www.utdallas.edu/~caldwell Prerequisites: One year of physical chemistry, one semester of advanced organic chemistry, or consent of instructor. Recommended Reference Text: “A Guide to Molecular Mechanics and Quantum Chemical Calculations,” Warren G. Hehre, Wavefunction, Inc., 2003, ISBN 189066118X Supplemental Reference texts: “Essentials of Computational Chemistry: Theories and Models”, Christopher J. Cramer, Wiley 2002, ISBN 0471485527 “Molecular Modeling and Simulation: an interdisciplinary guide”, Tamar Schlick, Springer 2002, ISBN 038795404X Spartan ’02 Windows: Tutorial and User’s Guide, Wavefunction Inc., 2001, ISBN 1890661198 “A Brief Guide to Molecular Mechanics and Quantum Chemical Calculations”, Warren J. Hehre, J. Yu, P.E. Klunzinger, and L. Lou, Wavefunction Inc. 1998, ISBN 1890661058 Recommended supply: USB pocket drive, 64Mbyte. Goals: The student will become familiar with the physical basis for molecular modeling and will learn to apply some of the currently available modeling packages to problems of interest in organic chemistry and structural biology. The emphasis will be on utility: How well do particular techniques reproduce reality and how much insight do they provide about chemistry? Software: Spartan, Gaussian 03, the Insight suite, VMD/NAMD (www.ks.uiuc.edu) Grading: There will be two homework assignments, comprising 30% of the grade. The two take-home exams will comprise 40%, and a project due on the last day of class will comprise 30%. -

NAMD User's Guide
NAMD User’s Guide Version 2.14 R. Bernardi, M. Bhandarkar, A. Bhatele, E. Bohm, R. Brunner, R. Buch, F. Buelens, H. Chen, C. Chipot, A. Dalke, S. Dixit, G. Fiorin, P. Freddolino, H. Fu, P. Grayson, J. Gullingsrud, A. Gursoy, D. Hardy, C. Harrison, J. H´enin, W. Humphrey, D. Hurwitz, A. Hynninen, N. Jain, W. Jiang, N. Krawetz, S. Kumar, D. Kunzman, J. Lai, C. Lee, J. Maia, R. McGreevy, C. Mei, M. Melo, M. Nelson, J. Phillips, B. Radak, J. Ribeiro, T. Rudack, O. Sarood, A. Shinozaki, D. Tanner, P. Wang, D. Wells, G. Zheng, F. Zhu August 5, 2020 Theoretical Biophysics Group University of Illinois and Beckman Institute 405 N. Mathews Urbana, IL 61801 Description The NAMD User’s Guide describes how to run and use the various features of the molecular dynamics program NAMD. This guide includes the capabilities of the program, how to use these capabilities, the necessary input files and formats, and how to run the program both on uniprocessor machines and in parallel. NAMD development is supported by National Institutes of Health grant NIH P41-GM104601. NAMD Version 2.14 Authors: R. Bernardi, M. Bhandarkar, A. Bhatele, E. Bohm, R. Brunner, R. Buch, F. Buelens, H. Chen, C. Chipot, A. Dalke, S. Dixit, G. Fiorin, P. Freddolino, H. Fu, P. Grayson, J. Gullingsrud, A. Gursoy, D. Hardy, C. Harrison, J. H´enin,W. Humphrey, D. Hurwitz, A. Hynninen, N. Jain, W. Jiang, N. Krawetz, S. Kumar, D. Kunzman, J. Lai, C. Lee, J. Maia, R. McGreevy, C. Mei, M. Melo, M. Nelson, J.