Ferritin Diversity: Mechanistic Studies, Disease Implications, and Materials Chemistry
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Importance of Eukaryotic Ferritins in Iron Handling and Cytoprotection
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archivio istituzionale della ricerca - Università di Brescia Biochem. J. (2015) 472, 1–15 doi:10.1042/BJ20150787 1 REVIEW ARTICLE The importance of eukaryotic ferritins in iron handling and cytoprotection Paolo Arosio*1, Fernando Carmona*, Raffaella Gozzelino†, Federica Maccarinelli* and Maura Poli* *Laboratory of Molecular Biology, Department of Molecular and Translational Medicine (DMMT), University of Brescia, Brescia, Italy †Inflammation and Neurodegeneration Laboratory, Chronic Disease Research Centre (CEDOC)/FCM, NOVA Medical School, Lisbon, Portugal Ferritins, the main intracellular iron storage proteins, have been structure is highly conserved among different phyla. It exerts an studied for over 60 years, mainly focusing on the mammalian important cytoprotective function against oxidative damage and ones. This allowed the elucidation of the structure of these proteins plays a role in innate immunity, where it also contributes to prevent and the mechanisms regulating their iron incorporation and parenchymal tissue from the cytotoxicity of pro-inflammatory mineralization. However, ferritin is present in most, although not agonists released by the activation of the immune response all, eukaryotic cells, comprising monocellular and multicellular activation. Less clear are the properties of the secretory ferritins invertebrates and vertebrates. The aim of this review is to provide expressed by insects and molluscs, which may be important for an update on the general properties of ferritins that are common to understanding the role played by serum ferritin in mammals. various eukaryotic phyla (except plants), and to give an overview on the structure, function and regulation of ferritins. -

Repression of Ferritin Light Chain Translation by Human Eif3
RESEARCH ARTICLE Repression of ferritin light chain translation by human eIF3 Mia C Pulos-Holmes1, Daniel N Srole1†, Maria G Juarez1, Amy S-Y Lee2, David T McSwiggen1, Nicholas T Ingolia1,3, Jamie H Cate1,3,4,5* 1Department of Molecular & Cell Biology, University of California, Berkeley, Berkeley, United States; 2Biology Department, Rosenstiel Basic Medical Science Research Center, Brandeis University, Waltham, United States; 3California Institute for Quantitative Biosciences, University of California, Berkeley, Berkeley, United States; 4Department of Chemistry, University of California, Berkeley, Berkeley, United States; 5Molecular Biophysics & Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, United States Abstract A central problem in human biology remains the discovery of causal molecular links between mutations identified in genome-wide association studies (GWAS) and their corresponding disease traits. This challenge is magnified for variants residing in non-coding regions of the genome. Single-nucleotide polymorphisms (SNPs) in the 5’ untranslated region (5’-UTR) of the ferritin light chain (FTL) gene that cause hyperferritinemia are reported to disrupt translation *For correspondence: FTL [email protected] repression by altering iron regulatory protein (IRP) interactions with the mRNA 5’-UTR. Here, we show that human eukaryotic translation initiation factor 3 (eIF3) acts as a distinct repressor of † Present address: Department FTL mRNA translation, and eIF3-mediated FTL repression is disrupted by a subset of SNPs in FTL of Molecular & Medical that cause hyperferritinemia. These results identify a direct role for eIF3-mediated translational Pharmacology, David Geffen control in a specific human disease. School of Medicine at UCLA, DOI: https://doi.org/10.7554/eLife.48193.001 University of California, Los Angeles, Los Angeles, United States Competing interests: The Introduction authors declare that no Iron is essential for a spectrum of metabolic pathways and cellular growth. -

Ferritin Heavy Chain (FTH1) Rabbit Polyclonal Antibody Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA326948 Ferritin Heavy Chain (FTH1) Rabbit Polyclonal Antibody Product data: Product Type: Primary Antibodies Applications: IF, IHC, WB Recommended Dilution: WB 1:500 - 1:2000, IHC 1:50- 1:200, IF 1:50- 1:200 Reactivity: Human, Mouse, Rat Host: Rabbit Isotype: IgG Clonality: Polyclonal Immunogen: Recombinant protein of human FTH1 Formulation: Store at -20C or -80C. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide, 50% glycerol, pH7.3 Concentration: lot specific Purification: Affinity purification Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Gene Name: ferritin heavy chain 1 Database Link: NP_002023 Entrez Gene 14319 MouseEntrez Gene 25319 RatEntrez Gene 2495 Human P02794 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 3 Ferritin Heavy Chain (FTH1) Rabbit Polyclonal Antibody – TA326948 Background: Ferritin (FTH) is a ubiquitous and highly conserved protein which plays a major role in iron homeostasis by sequestering and storing iron in a non-toxic and bioavailable form. The assembled ferritin molecule, often referred to as a nanocage, can store up to 4,500 atoms of iron. It forms a holoenzyme of ~450 kDa, consisting of 24 subunits made up of two types of polypeptide chains: ferritin heavy chain and ferritin light chain, each having unique functions. -

Ferritin Heavy Chain (F23): Sc-51887
SANTA CRUZ BIOTECHNOLOGY, INC. ferritin heavy chain (F23): sc-51887 The Power to Question BACKGROUND APPLICATIONS Mammalian ferritins consist of 24 subunits made up of two types of polypep- ferritin heavy chain (F23) is recommended for detection of ferritin of human tide chains, ferritin heavy chain and ferritin light chain, which each have origin by ELISA. unique functions. Ferritin heavy chains catalyze the first step in iron storage, Suitable for use as control antibody for ferritin heavy chain siRNA (h): the oxidation of Fe (II), whereas ferritin light chains promote the nucleation sc-40575. of ferrihydrite, enabling storage of Fe (III). The most prominent role of mam- malian ferritins is to provide iron-buffering capacity to cells. In addition to iron Molecular Weight of ferritin heavy chain: 21 kDa. buffering, heavy chain ferritin is also involved in the regulation of thymidine biosynthesis via increased expression of cytoplasmic serine hydroxymethyl- STORAGE transferase, which is a limiting factor in thymidylate synthesis in MCF-7 cells. Store at 4° C, **DO NOT FREEZE**. Stable for one year from the date of Light chain ferritin is involved in cataracts by at least two mechanisms, shipment. Non-hazardous. No MSDS required. hereditary hyperferritinemia cataract syndrome, in which light chain ferritin is overexpressed, and oxidative stress, an important factor in the develop- PROTOCOLS ment of ageing-related cataracts. The gene encoding human ferritin heavy See our web site at www.scbt.com or our catalog for detailed protocols chain maps to chromosome 11q13 and the human ferritin light chain gene and support products. maps to chromosome 19q13.3-q13.4. -

Ferritin L Antibody
Efficient Professional Protein and Antibody Platforms Ferritin L Antibody Basic information: Catalog No.: UPA63379 Source: Rabbit Size: 50ul/100ul Clonality: Monoclonal Concentration: 1mg/ml Isotype: Rabbit IgG Purification: Protein A affinity purified Useful Information: WB:1:1000-1:5000 Applications: IHC:1:100-1:500 ICC:1:100-1:200 Reactivity: Human, Mouse, Rat Specificity: This antibody recognizes Ferritin L protein. Immunogen: Recombinant protein Mammalian ferritins consist of 24 subunits made up of two types of poly- peptide chains, ferritin heavy chain and ferritin light chain, which each have unique functions. Ferritin heavy chains catalyze the first step in iron storage, the oxidation of Fe (II), whereas ferritin light chains promote the nucleation of ferrihydrite, enabling storage of Fe (III). The most prominent role of mammalian ferritins is to provide iron-buffering capacity to cells. In addition to iron buffering, heavy chain ferritin is also involved in the regulation of Description: thymidine biosynthesis via increased expression of cytoplasmic serine hy- droxymethyltransferase, which is a limiting factor in thymidylate synthesis in MCF-7 cells. Light chain ferritin is involved in cataracts by at least two mechanisms, hereditary hyperferritinemia cataract syndrome, in which light chain ferritin is overexpressed, and oxidative stress, an important factor in the development of ageing-related cataracts. The gene encoding human ferritin heavy chain maps to chromosome 11q13 and the human ferritin light chain gene maps to chromosome 19q13.3-q13.4. Uniprot: P02792(Human) P29391(Mouse) P02793(Rat) BiowMW: 54 kDa Buffer: 1*TBS (pH7.4), 1%BSA, 40%Glycerol. Preservative: 0.05% Sodium Azide. Storage: Store at 4°C short term and -20°C long term. -

(12) United States Patent (10) Patent No.: US 8,084,017 B2 Ahrens (45) Date of Patent: Dec
US008O84017B2 (12) United States Patent (10) Patent No.: US 8,084,017 B2 Ahrens (45) Date of Patent: Dec. 27, 2011 (54) CONTRASTAGENTS FOR MAGNETIC Cohen, B., et al., “Ferritin as an Endogenous MRI Reporter for RESONANCE MAGING AND METHODS Noninvasive Imaging of Gene Expression in C6 Glioma Tumors.” RELATED THERETO Neoplasia, 7(2):109-117 (2005). Genove, G., “A new transgene reporter for in vivo magnetic reso (75) Inventor: Eric Ahrens, Pittsburgh, PA (US) nance imaging.” Nature Medicine, 11(4):450-454 (2005). Ichikawa, T., et al., “MRI of Transgene Expression: Correlation of (73) Assignee: Carnegie Mellon University, Pittsburgh, Therapeutic Gene Expression.” Neoplasia, 4(6):523-530 (2002). PA (US) Moore, A., et al., “Measuring transferrin receptor gene expression by NMR imaging.” Biochimica et Biophysica Acta, 14 02:239-249 (*) Notice: Subject to any disclaimer, the term of this (1998). patent is extended or adjusted under 35 Walter, G., et al., “Noninvasive measurement of gene expression in U.S.C. 154(b) by 738 days. skeletal muscle.” PNAS,97(10):5151-5155 (2000). Weissleder, R., et al., “MR Imaging and Scintigraphy of Gene (21) Appl. No.: 10/384,496 Expression through Melanin Induction.” Radiology, 204:425-429 (1997). (22) Filed: Mar. 7, 2003 Ahrens, et al., “A model for MRI contrast enhancement using T. agents”. Proc. Acad. Sci. USA. vol. 95, pp. 8443-8448, Applied (65) Prior Publication Data Biological Sciences (1998). Ahrens, et al., “Peripheral somatosensory fMRI in mouse at 11.7T". US 2003/0219385 A1 Nov. 27, 2003 NMR in Biomedicine, NMR Biomed. 14:318-324 (2001). -

Iron Loading-Induced Aggregation and Reduction of Iron Incorporation in Heteropolymeric Ferritin Containing a Mutant Light Chain That Causes Neurodegeneration
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1812 (2011) 544–548 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Iron loading-induced aggregation and reduction of iron incorporation in heteropolymeric ferritin containing a mutant light chain that causes neurodegeneration Barry B. Muhoberac a,⁎,1, Martin A. Baraibar b,1, Ruben Vidal b,⁎ a Department of Chemistry and Chemical Biology, Indiana University–Purdue University, Indianapolis, IN 46202, USA b Department of Pathology and Laboratory Medicine, Indiana University School of Medicine, Indianapolis, IN, 46202, USA article info abstract Article history: Hereditary ferritinopathy (HF) is a neurodegenerative disease characterized by intracellular ferritin inclusion Received 18 June 2010 bodies (IBs) and iron accumulation throughout the central nervous system. Ferritin IBs are composed of Received in revised form 14 October 2010 mutant ferritin light chain as well as wild-type light (Wt-FTL) and heavy chain (FTH1) polypeptides. In vitro Accepted 18 October 2010 studies have shown that the mutant light chain polypeptide p.Phe167SerfsX26 (Mt-FTL) forms soluble ferritin Available online 26 October 2010 24-mer homopolymers having a specific structural disruption that explains its functional problems of reduced ability to incorporate iron and aggregation during iron loading. However, because ferritins are usually 24-mer Keywords: Ferritin heteropolymers and all three polypeptides are found in IBs, we investigated the properties of Mt-FTL/FTH1 and Iron Mt-FTL/Wt-FTL heteropolymeric ferritins. We show here the facile assembly of Mt-FTL and FTH1 subunits into Pore soluble ferritin heteropolymers, but their ability to incorporate iron was significantly reduced relative to Wt- Neurodegeneration FTL/FTH1 heteropolymers. -

Ferritin L-Subunit Gene Mutation and Hereditary Hyperferritinaemia Cataract Syndrome: a Case Report and Literature Review
Ferritin L-subunit Gene Mutation and Hereditary Hyperferritinaemia Cataract Syndrome: A Case Report and Literature Review Yunfan Yang Sichuan University West China Hospital Ting Lin Sichuan University West China Hospital Xincuan Chen ( [email protected] ) Sichuan University West China Hospital https://orcid.org/0000-0001-9292-9623 Case Report Keywords: L-Ferritin, cataract, gene, mutation Posted Date: March 9th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-290096/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/16 Abstract Background: Hereditary hyperferritinaemia cataract syndrome (HHCS) is an autosomal dominant disease characterized by high serum ferritin levels and juvenile bilateral cataracts. It is often caused by mutations in the Iron Response Element (IRE) of the ferritin L-subunit (FTL) gene. Most of the mutations are point mutations located in the upper stem and the conserved hexanucleotide of the hairpin structure of IRE, only a few mutations are deletions. Case presentation: Here we report a 73-year-old woman who presented to clinic with persistently elevated serum ferritin and family history of juvenile bilateral cataracts in four generations. DNA sequencing analyses identied a heterozygous c.-167C>T mutation in the 5’ untranslated region (UTR) of the FTL gene. Her daughter and granddaughter were also conrmed to have the same genetic mutation. Conclusion: HHCS should be considered in the differential diagnosis of hyperferritinemia, especially in the presence of normal serum iron concentration and transferrin saturation. For patients with unexplained hyperferritinemia and bilateral cataracts who have experienced early vision loss, the establishment of genetic counseling is essential to diagnose other family members who are at risk in time, so as to avoid unnecessary liver biopsy and venesection. -

Screening and Identification of Endometrial Proteins As Novel Potential Biomarkers for Repeated Implantation Failure
Screening and identification of endometrial proteins as novel potential biomarkers for repeated implantation failure Chong Wang1,2, Ying Feng1, Wen-Jing Zhou1, Zhao-Jun Cheng1, Mei-Yan Jiang1, Yan Zhou1 and Xiao-Yang Fei1 1 Hangzhou Women's Hospital (Hangzhou Maternity and Child Health Care Hospital), Hangzhou, China 2 Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine, Hangzhou, China ABSTRACT Inadequate endometrial receptivity may be responsible for the low implantation rate of transferred embryos in in vitro fertilization (IVF) treatments. Patients with repeated implantation failure (RIF) impact the clinical pregnancy rate for IVF. We collected endometrial tissue during the implantation window of hysteroscopy biopsies from September 2016 to December 2019 and clinical data were collected simultaneously. Patients were divided into RIF and pregnant controls group according to pregnancy outcomes. A total of 82 differentially expressed endometrial proteins were identified, including 55 up-regulated proteins (>1.50-fold, P < 0:05) and 27 down-regulated proteins (<0.67-fold, P < 0:05) by iTRAQ labeling coupled with the 2D LC MS/MS technique in the RIF group. String analysis found interactions between these proteins which assembled in two bunches: ribosomal proteins and blood homeostasis proteins. The most significant enriched Gene Ontology terms were negative regulation of hydrolase activity, blood microparticle, and enzyme inhibitor activity. Our results emphasized the corticosteroid-binding globulin and -

Hereditary Hyperferritinemia Cataract Syndrome: Ferritin L Gene and Physiopathology Behind the Disease—Report of New Cases
International Journal of Molecular Sciences Case Report Hereditary Hyperferritinemia Cataract Syndrome: Ferritin L Gene and Physiopathology behind the Disease—Report of New Cases Ferran Celma Nos 1,† , Gonzalo Hernández 1,†,Xènia Ferrer-Cortès 1,2, Ines Hernandez-Rodriguez 3, Begoña Navarro-Almenzar 4 , José Luis Fuster 5 , Mar Bermúdez Cortés 5, Santiago Pérez-Montero 2, Cristian Tornador 2 and Mayka Sanchez 1,2,* 1 Iron Metabolism: Regulation and Diseases, Department of Basic Sciences, Universitat Internacional de Catalunya (UIC), 08195 Sant Cugat del Vallès, Spain; [email protected] (F.C.N.); [email protected] (G.H.); [email protected] (X.F.-C.) 2 BloodGenetics S.L. Diagnostics in Inherited Blood Diseases, 08950 Esplugues de Llobregat, Spain; [email protected] (S.P.-M.); [email protected] (C.T.) 3 Hematology Service, University Hospital Germans Trias i Pujol (HGTiP), Institut Català d’Oncología (ICO), 08916 Badalona, Spain; [email protected] 4 Hematology and Hemotherapy Service, Clinic University Hospital Virgen de la Arrixaca, Instituto Murciano de Investigación Biosanitaria (IMIB), 30120 Murcia, Spain; [email protected] 5 Pediatric OncoHematology Service, Clinic University Hospital Virgen de la Arrixaca, Instituto Murciano de Investigación Biosanitaria (IMIB), 30120 Murcia, Spain; [email protected] (J.L.F.); [email protected] (M.B.C.) Citation: Celma Nos, F.; Hernández, * Correspondence: [email protected] G.; Ferrer-Cortès, X.; † These authors contributed equally to this work. Hernandez-Rodriguez, I.; Navarro-Almenzar, B.; Fuster, J.L.; Abstract: Hereditary hyperferritinemia-cataract syndrome (HHCS) is a rare disease characterized by Bermúdez Cortés, M.; Pérez-Montero, high serum ferritin levels, congenital bilateral cataracts, and the absence of tissue iron overload. -
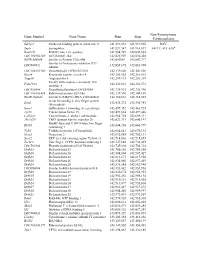
Gene Symbol Gene Name Start Stop Non-Synonymous Polymorphism
Non-Synonymous Gene Symbol Gene Name Start Stop Polymorphism Sdcbp2 Syndecan binding protein (syntenin) 2 141,891,232 141,919,905 I82V Snph Syntaphilin 141,921,547 141,961,697 A411T, 415_420* Rad21l1 RAD21-like 1 (S. pombe) 141,984,729 142,009,665 LOC100363280 mCG140681-like 142,022,959 142,030,458 RGD1564048 Similar to Protein C20orf46 142,043961 142,047,717 Similar to Proteasome inhibitor PI31 LOC689852 142,058,695 142,083,908 subunit LOC100363380 Hypothetical LOC100363380 142,139,020 142,143,826 Rspo4 R-spondin family, member 4 142,185,028 142,216,015 Angpt4 Angiopoietin 4 142,249,115 142,282,307 Family with sequence similarity 110, Fam110a 142,326,013 142,328,572 member A LOC689884 Hypothetical protein LOC689884 142,335,923 142,336,396 LOC100363434 Ribosomal protein S29-like 142,339,596 142,341,169 RGD1304644 Similar to RIKEN cDNA 2310046K01 142,360,661 142,365,285 Scratch homolog 2, zinc finger protein Scrt2 142,434,272 142,445,984 (Drosophila) Srxn1 Sulfiredoxin 1 homolog (S. cerevisiae) 142,457,352 142,462,725 Tcf15 Transcription factor 15 142,491,664 142,497,446 Csnk2a1 Casein kinase 2, alpha 1 polypeptide 142,564,754 142,609,311 Tbc1d20 TBC1 domain family, member 20 142,623,713 142,640,197 RanBP-type and C3HC4-type zinc finger Rbck1 142,644,156 142,660,799 containing 1 Trib3 Tribbles homolog 3 (Drosophila) 142,664,641 142,670,135 Nrsn2 Neurensin 2 142,692,988 142,702,131 Sox12 SRY (sex determining region Y)-box 12 142,714,836 142,715,855 Zcchc3 Zinc finger, CCHC domain containing 3 142,727,648 142,730,465 LOC502684 Hypothetical -

Title Biology of Ferritin in Mammals: an Update on Iron Storage, Oxidative Damage and Neurodegeneration. Dario Finazzi , MD
Title Biology of ferritin in mammals: an update on iron storage, oxidative damage and neurodegeneration. Dario Finazzi1,2, MD and Paolo Arosio, PhD1 1 Department of Molecular and Translational Medicine, University of Brescia, Viale Europa 11, 25123 Brescia, Italy 2 Spedali Civili di Brescia, P.le Spedali Civili, 1, 25123 Brescia, Italy Corresponding Author Dario Finazzi Department of Molecular and Translational Medicine, University of Brescia, Viale Europa 11, 25123 Brescia, Italy Phone ++390303717334 Fax ++390303717305 Email [email protected] Abstract Iron is an abundant transition metal that is essential for life, being associated to many enzyme and oxygen carrier proteins involved in a variety of fundamental cellular processes. At the same time the metal is potentially toxic due to its capacity to engage in the catalytic production of noxious reactive oxygen species. The control of iron availability in the cells is largely dependent on ferritins, ubiquitous proteins with storage and detoxification capacity. In mammals, cytosolic ferritins are composed of two types of subunits, the H and the L chain, assembled to form a 24-mer spherical cage. Ferritin is present also in mitochondria, in the form of a complex with 24 identical chains. Even though the proteins have been known for a long time, their study is a very active and interesting field yet. In this review we will focus our attention to mammalian cytosolic and mitochondrial ferritins, describing the most recent advancement regarding their storage and anti- oxidant function, the effects of their genetic mutations in human pathology, and also the possible involvement in non-iron related activities.