Modeling Down Syndrome Neurodevelopment with Dosage Compensation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

Klinefelter Syndrome) at 9 Years of Age
17th SSBP International Research Symposium Developmental trajectories of behavioural phenotypes Programme Book 10–13 October 2014 • New York, USA 2 17th International SSBP Research Symposium The Society for the Study 3 of Behavioural Phenotypes 10th – 13th October 2014 The 17th SSBP International Meeting Developmental Trajectories of Behavioural Phenotypes New York, USA 10th – 13th October 2014, New York, USA Contents Contents Welcome ........................................................................................................................................................................................................9 New York Conference Organiser ..............................................................................................................................................10 Scientific Committee ..........................................................................................................................................................................11 4 The SSBP .......................................................................................................................................................................................................12 The SSBP Executive Committee : ...........................................................................................................................................................................12 Meetings of the SSBP ....................................................................................................................................................................................................13 -

A Simple Differentiation Protocol for Generation of Induced
1 Article, Special Issue "Induced Pluripotent Stem Cells in Neurodegenerative Diseases: Application for Therapy and 2 Disease Modeling" 3 A simple differentiation protocol for generation of 4 induced pluripotent stem cell-derived basal forebrain 5 cholinergic neurons for Alzheimer’s disease and 6 frontotemporal dementia disease modeling 7 Supplemental information 8 Method 1. Reprogramming and characterisation of MBE2960 healthy control iPSC line 9 The iPSCs were generated using skin fibroblasts obtained from subjects over the age of 18 years 10 by episomal method as described [40]. Briefly, reprogramming was performed on passage 8-10 11 fibroblasts by nucleofection (Lonza Amaxa Nucleofector) with episomal vectors expressing 12 OCT4, SOX2, KLF4, L-MYC, LIN28 and shRNA against p53 [41] in feeder- and serum- free 13 conditions using TeSR-E7 medium (Stemcell Technologies). Subsequently, reprogrammed 14 colonies were manually dissected to establish clonal cell lines [42]. Three clones were assessed 15 for pluripotency markers via immunocytochemistry (Figure S1A). The iPSC line was expanded 16 and characterised. Embryoid bodies were obtained as described [43] and using tri-lineage 17 differentiation kit (Stemcell Technologies). Germ layer differentiation was assessed by 18 immunochemistry (Figure S1B). Copy number variation (CNV) analysis of original fibroblasts 19 and iPSCs from MBE2960 (Figure S1C) was performed using Illumina HumanCore Beadchip 20 arrays as we described [40]. CNV analyses were performed using PennCNV and QuantiSNP with 21 default parameter settings [44,45]. Chromosomal aberrations were deemed to involve at least 10 22 contiguous single nucleotide polymorphisms (SNPs) or a genomic region spanning at least 1MB 23 [44,45]. The B allele frequency (BAF) and the log R ratio (LRR) were extracted from 24 GenomeStudio (Illumina) for representation (Figure S1D). -

Diagnosis of FOXG1 Syndrome Caused by Recurrent Balanced Chromosomal Rearrangements: Case Study and Literature Review Connor P
Craig et al. Mol Cytogenet (2020) 13:40 https://doi.org/10.1186/s13039-020-00506-1 CASE REPORT Open Access Diagnosis of FOXG1 syndrome caused by recurrent balanced chromosomal rearrangements: case study and literature review Connor P. Craig1,2, Emily Calamaro3, Chin‑To Fong3, Anwar M. Iqbal1, Alexander R. Paciorkowski3,4,5,6 and Bin Zhang1,3* Abstract Background: The FOXG1 gene plays a vital role in mammalian brain diferentiation and development. Intra‑ and intergenic mutations resulting in loss of function or altered expression of the FOXG1 gene cause FOXG1 syndrome. The hallmarks of this syndrome are severe developmental delay with absent verbal language, post‑natal growth restric‑ tion, post‑natal microcephaly, and a recognizable movement disorder characterized by chorea and dystonia. Case presentation: Here we describe a case of a 7‑year‑old male patient found to have a de novo balanced translo‑ cation between chromosome 3 at band 3q14.1 and chromosome 14 at band 14q12 via G‑banding chromosome and Fluorescence In Situ Hybridization (FISH) analyses. This rearrangement disrupts the proximity of FOXG1 to a previously described smallest region of deletion overlap (SRO), likely resulting in haploinsufciency. Conclusions: This case adds to the growing body of literature implicating chromosomal structural variants in the manifestation of this disorder and highlights the vital role of cis‑acting regulatory elements in the normal expression of this gene. Finally, we propose a protocol for refex FISH analysis to improve diagnostic efciency for patients with suspected FOXG1 syndrome. Keywords: FOXG1, Haploinsufciency, Postnatal microcephaly, FISH, Enhancer, Chromosomal rearrangement, Diagnosis Introduction brain development, with high levels of expression in the Te Forkhead Box G1 (FOXG1) gene [OMIM: 164874], developing fetal telencephalon [1–4]. -

Notch Signal Controls Several Steps of Inner Ear Development Norio Yamamoto and Matthew W
NIDCD Notch signal controls several steps of inner ear development Norio Yamamoto and Matthew W. Kelley Section on Developmental Neuroscience Section on Developmental Neuroscience National Institute on Deafness and Other Communication Disorders National Institutes of Health Abstract Problem addressed RBP-J mutant cochleae did not have any supporting cells Notch signaling has been reported to contribute to inner ear development, however, its specific functions remain unclear, partly because of discrepancies between the phenotypes of mutant mice with single deletion of specific Notch related genes. These discrepancies are probably due to functional compensation by other Notch Figure 4 receptors or ligands. Foxg1 Cre;RBP-J floxed/+ Foxg1 Cre;RBP-J floxed/floxed A B C D To determine the effects of complete elimination of Notch signaling, we used a conditional knockout of the Rbpsuh gene. RBP-J protein is a critical transcriptional p27 Phalloidin p27 Phalloidin To test if mutant cochleae contained supporting cells we co-activator for all Notch molecules and thus deletion of this protein inhibits all Notch signaling. examined expression of supporting cell markers such as p27 and Prox1. On E17.5 p27 was expressed in supporting cells Methods and Measures ***** ***** under or between hair cells such as inner pharyngeal cells, Floxed Rbpsuh mice were crossed with Foxg1-Cre knock-in mice to delete the Rbpsuh gene in the inner ear. Inner ear phenotypes in Rbpsuh conditional knockout Deiter's cells and pillar cells (asterisks in figure 4 A and B). mice were determined at various developmental stages using immunohistochemistry. But no p27 expression was detected in those regions of RBP-J E Prox1 F Phalloidin G Prox1 H Phalloidin conditional knockout mice (Figure 4 C and D). -

Aberrant Expression of Enzymes Regulating M6a Mrna Methylation: Implication in Cancer
Cancer Biol Med 2018. doi: 10.20892/j.issn.2095-3941.2018.0365 REVIEW Aberrant expression of enzymes regulating m6A mRNA methylation: implication in cancer Natalia Pinello1,2, Stephanie Sun1,2, Justin Jong-Leong Wong1,2 1Epigenetics and RNA Biology Program Centenary Institute, The University of Sydney, Camperdown 2050, Australia; 2Sydney Medical School, The University of Sydney, Camperdown 2050, Australia ABSTRACT N6-methyladenosine (m6A) is an essential RNA modification that regulates key cellular processes, including stem cell renewal, cellular differentiation, and response to DNA damage. Unsurprisingly, aberrant m6A methylation has been implicated in the development and maintenance of diverse human cancers. Altered m6A levels affect RNA processing, mRNA degradation, and translation of mRNAs into proteins, thereby disrupting gene expression regulation and promoting tumorigenesis. Recent studies have reported that the abnormal expression of m6A regulatory enzymes affects m6A abundance and consequently dysregulates the expression of tumor suppressor genes and oncogenes, including MYC, SOCS2, ADAM19, and PTEN. In this review, we discuss the specific roles of m6A “writers", “erasers”, and “readers” in normal physiology and how their altered expression promotes tumorigenesis. We also describe the potential of exploiting the aberrant expression of these enzymes for cancer diagnosis, prognosis, and the development of novel therapies. KEYWORDS RNA modification; N6-methyladenosine (m6A); cancer; tumor suppressor; oncogene Introduction mRNAs and their consequent transcriptional outcomes include RNA specific methylases (writers), demethylases RNA modifications have recently been shown to play (erasers), and reader proteins (Figure 1). important roles in normal and disease biology. Over 170 Together, the tightly-regulated functions of m6A writers, different types of post-transcriptional modifications have erasers, and readers are critical in maintaining the integrity of been identified in RNA, many of which have unknown m6A RNA modification in cells. -

Orphanet Report Series Rare Diseases Collection
Marche des Maladies Rares – Alliance Maladies Rares Orphanet Report Series Rare Diseases collection DecemberOctober 2013 2009 List of rare diseases and synonyms Listed in alphabetical order www.orpha.net 20102206 Rare diseases listed in alphabetical order ORPHA ORPHA ORPHA Disease name Disease name Disease name Number Number Number 289157 1-alpha-hydroxylase deficiency 309127 3-hydroxyacyl-CoA dehydrogenase 228384 5q14.3 microdeletion syndrome deficiency 293948 1p21.3 microdeletion syndrome 314655 5q31.3 microdeletion syndrome 939 3-hydroxyisobutyric aciduria 1606 1p36 deletion syndrome 228415 5q35 microduplication syndrome 2616 3M syndrome 250989 1q21.1 microdeletion syndrome 96125 6p subtelomeric deletion syndrome 2616 3-M syndrome 250994 1q21.1 microduplication syndrome 251046 6p22 microdeletion syndrome 293843 3MC syndrome 250999 1q41q42 microdeletion syndrome 96125 6p25 microdeletion syndrome 6 3-methylcrotonylglycinuria 250999 1q41-q42 microdeletion syndrome 99135 6-phosphogluconate dehydrogenase 67046 3-methylglutaconic aciduria type 1 deficiency 238769 1q44 microdeletion syndrome 111 3-methylglutaconic aciduria type 2 13 6-pyruvoyl-tetrahydropterin synthase 976 2,8 dihydroxyadenine urolithiasis deficiency 67047 3-methylglutaconic aciduria type 3 869 2A syndrome 75857 6q terminal deletion 67048 3-methylglutaconic aciduria type 4 79154 2-aminoadipic 2-oxoadipic aciduria 171829 6q16 deletion syndrome 66634 3-methylglutaconic aciduria type 5 19 2-hydroxyglutaric acidemia 251056 6q25 microdeletion syndrome 352328 3-methylglutaconic -

Downloaded from Bioscientifica.Com at 09/26/2021 08:38:14PM Via Free Access
1 185 L C Gregory, P Gergics, OTX2 mutations in congenital 185:1 121–135 Clinical Study M Nakaguma and others hypopituitarism The phenotypic spectrum associated with OTX2 mutations in humans Louise C Gregory 1,*, Peter Gergics2,* , Marilena Nakaguma3,* , Hironori Bando2 , Giuseppa Patti1,4,5, Mark J McCabe1, Qing Fang 2, Qianyi Ma2 , Ayse Bilge Ozel2 , Jun Z Li2 , Michele Moreira Poina3, Alexander A L Jorge 3, Anna F Figueredo Benedetti3, Antonio M Lerario3, Ivo J P Arnhold3 , Berenice B Mendonca3 , Mohamad Maghnie4,5, Sally A Camper2 , Luciani R S Carvalho3 and Mehul T Dattani1 1Section of Molecular Basis of Rare Disease, Genetics and Genomic Medicine Research & Teaching Department, UCL Great Ormond Street Institute of Child Health, London, UK, 2Department of Human Genetics, University of Michigan, Correspondence Ann Arbor, Michigan, USA, 3Developmental Endocrinology Unit, Hospital das Clinicas da Faculdade de Medicina da should be addressed Universidade de São Paulo, São Paulo, Brazil, 4Department of Pediatrics, IRCCS Istituto Giannina Gaslini and to S A Camper or M T 5Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Dattani Genova, Genova, Italy Email *(L C Gregory, P Gergics and M Nakaguma contributed equally to this work) [email protected] or [email protected] Abstract Objective: The transcription factor OTX2 is implicated in ocular, craniofacial, and pituitary development. Design: We aimed to establish the contribution of OTX2 mutations in congenital hypopituitarism patients with/without eye abnormalities, study functional consequences, and establish OTX2 expression in the human brain, with a view to investigate the mechanism of action. Methods: We screened patients from the UK (n = 103), international centres (n = 24), and Brazil (n = 282); 145 were within the septo-optic dysplasia spectrum, and 264 had no eye phenotype. -
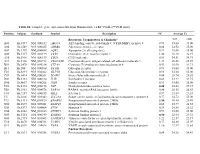
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90 -

Inside This Issue
Winter 2014 No. 77 Inside this issue Group News | Fundraising | Members’ Letters | One Family Living with Two Different Chromosome Disorders | Bristol Conference 2014 | Unique Leaflets | Christmas Card Order Form Sophie, Unique’s Chair of Trustees Dear Members, In the past month a few things have reminded me of why it is so important to make connections through Unique but also to draw support from other parents around us. I’ve just returned from Unique’s most recent family conference in Bristol where 150 of us parents and carers had a lovely time in workshops, meals and activities, chatting and watching our children milling around together like one big family since – although we had never met before – we have shared so many experiences in common. However in contrast I have also just met a new mum who has just moved to my area from far away with two toddlers, one with a rare joys of the internet, it is becoming easier to meet others with similar, chromosome disorder, who is starting from scratch with no even very rare, chromosome disorders around the world and to find professional, medical or social support. She reminds me of how yourself talking to them in the middle of the night about some lonely I felt when Max was newly diagnosed, when I knew no one interesting things our children share in common (obsession with with a disabled child let alone anyone with a rare chromosome catalogues, anyone?) And of course we also have an enormous disorder. Elsewhere our latest Unique Facebook group, Unique amount in common with so many parents of children with other Russia, is also just starting up – so far it includes just a small special needs or disabilities around us in our own communities who number of members sharing very different experiences to mine here will often be walking the same path as us. -

RBM15 Modulates the Function of Chromatin Remodeling Factor BAF155 Through RNA Methylation in Developing Cortex
Molecular Neurobiology (2019) 56:7305–7320 https://doi.org/10.1007/s12035-019-1595-1 RBM15 Modulates the Function of Chromatin Remodeling Factor BAF155 Through RNA Methylation in Developing Cortex Yuanbin Xie1,2 & Ricardo Castro-Hernández1 & Godwin Sokpor1 & Linh Pham1 & Ramanathan Narayanan1,3 & Joachim Rosenbusch1 & Jochen F. Staiger1,2 & Tran Tuoc1,2 Received: 5 February 2019 /Accepted: 2 April 2019 /Published online: 24 April 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Chromatin remodeling factor BAF155 is an important regulator of many biological processes. As a core and scaffold subunit of the BAF (SWI/SNF-like) complex, BAF155 is capable of regulating the stability and function of the BAF complex. The spatiotemporal expression of BAF155 during embryogenesis is essential for various aspects of organogenesis, particularly in the brain development. However, our understanding of the mechanisms that regulate the expression and function of BAF155 is limited. Here, we report that RBM15, a subunit of the m6A methyltransferase complex, interacts with BAF155 mRNA and mediates BAF155 mRNA degradation through the mRNA methylation machinery. Ablation of endogenous RBM15 expression in cultured neuronal cells and in the developing cortex augmented the expression of BAF155. Conversely, RBM15 overexpres- sion decreased BAF155 mRNA and protein levels, and perturbed BAF155 functions in vivo, including repression of BAF155- dependent transcriptional activity and delamination of apical radial glial progenitors as a hallmark of basal radial glial progenitor genesis. Furthermore, we demonstrated that the regulation of BAF155 by RBM15 depends on the activity of the mRNA methylation complex core catalytic subunit METTL3. Altogether, our findings reveal a new regulatory avenue that elucidates how BAF complex subunit stoichiometry and functional modulation are achieved in mammalian cells. -

Cortical Foxp2 Supports Behavioral Flexibility and Developmental Dopamine D1 Receptor Expression
bioRxiv preprint doi: https://doi.org/10.1101/624973; this version posted May 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Cortical Foxp2 supports behavioral flexibility and developmental dopamine D1 receptor expression Marissa Co, Stephanie L. Hickey, Ashwinikumar Kulkarni, Matthew Harper, Genevieve Konopka* Department of Neuroscience, University of Texas Southwestern Medical Center, Dallas, TX, USA *Correspondence: [email protected] Contact information Genevieve Konopka, Ph.D. Department of Neuroscience, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., ND4.300, Dallas, TX 75390-9111 TEL: 214-648-5135, FAX: 214-648-1801, Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/624973; this version posted May 2, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Abstract FoxP2 encodes a forkhead box transcription factor required for the development of neural circuits underlying language, vocalization, and motor-skill learning. Human genetic studies have associated FOXP2 variation with neurodevelopmental disorders (NDDs), and within the cortex, it is coexpressed and interacts with other NDD-associated transcription factors. Cortical Foxp2 is required in mice for proper social interactions, but its role in other NDD-relevant behaviors and molecular pathways is unknown.