An Integrative Framework for Bayesian Variable Selection with Informative Priors for Identifying Genes and Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Analyses of Human Fetal Retinal Pigment Epithelium Gene Expression Suggest Ocular Disease Mechanisms
ARTICLE https://doi.org/10.1038/s42003-019-0430-6 OPEN Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms Boxiang Liu 1,6, Melissa A. Calton2,6, Nathan S. Abell2, Gillie Benchorin2, Michael J. Gloudemans 3, 1234567890():,; Ming Chen2, Jane Hu4, Xin Li 5, Brunilda Balliu5, Dean Bok4, Stephen B. Montgomery 2,5 & Douglas Vollrath2 The retinal pigment epithelium (RPE) serves vital roles in ocular development and retinal homeostasis but has limited representation in large-scale functional genomics datasets. Understanding how common human genetic variants affect RPE gene expression could elu- cidate the sources of phenotypic variability in selected monogenic ocular diseases and pin- point causal genes at genome-wide association study (GWAS) loci. We interrogated the genetics of gene expression of cultured human fetal RPE (fRPE) cells under two metabolic conditions and discovered hundreds of shared or condition-specific expression or splice quantitative trait loci (e/sQTLs). Co-localizations of fRPE e/sQTLs with age-related macular degeneration (AMD) and myopia GWAS data suggest new candidate genes, and mechan- isms by which a common RDH5 allele contributes to both increased AMD risk and decreased myopia risk. Our study highlights the unique transcriptomic characteristics of fRPE and provides a resource to connect e/sQTLs in a critical ocular cell type to monogenic and complex eye disorders. 1 Department of Biology, Stanford University, Stanford, CA 94305, USA. 2 Department of Genetics, Stanford University School of Medicine, Stanford, CA 94305, USA. 3 Program in Biomedical Informatics, Stanford University School of Medicine, Stanford 94305 CA, USA. 4 Department of Ophthalmology, Jules Stein Eye Institute, UCLA, Los Angeles 90095 CA, USA. -

SUPPLEMENTARY MATERIALS and METHODS Mouse Strains
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut SUPPLEMENTARY MATERIALS AND METHODS Mouse strains, diethylnitrosamine (DEN) treatments, diets, animals care Mouse strains – Liver-specific PTEN knockout mice (C57BL/6J, AlbCre/Ptenflox/flox, LPTENKO) and Ptenflox/flox (CTL, littermates of LPTENKO) were previously described[1]. Liver-specific inducible PTEN knockout mice (AlbCre-ERT2Tg/+/Ptenflox/flox, LIPTENKO) were generated by crossing AlbCre-ERT2Tg/+ with Ptenflox/flox mice. Two-months old LIPTENKO were injected with tamoxifen to induce PTEN deletion in hepatocytes and analyzed either 9 days or 3 months post-deletion. db/db and control mice were obtained from Charles River Laboratories (C57BLKS/J). Liver samples from ob/ob and control mice (B6. V-Lepob/JRj) were provided by Pr. Jeanrenaud-Rohner (Geneva University). Diethylnitrosamine treatments – For Diethylnitrosamine (DEN)-induced HCC models, 15 days-old mice (C57BL/6J) were injected intraperitoneally with 25mg/kg of DEN (Sigma-Aldrich). Animals were sacrificed 11 months post-injection. Blood/tissues samples were collected and stored at -80°C. For acute DEN treatment (tumor initiation), 2-months old male mice (C57BL/6J) were injected intraperitoneally with 100mg/kg of DEN and sacrificed 48h after. Methionine-Choline Deficient Diet – 8 weeks-old C57BL/6J mice were fed with a standard or methionine-choline deficient diet (MCD; ssniff Spezialdiäten GmbH, Soest, Germany) (22 kJ% fat, 14KJ% protein, 64 KJ% carbohydrates), for 2 weeks. At the end of the study, mice were anesthetized by isoflurane and sacrificed by decapitation and blood and organs were collected and weighted. -

Genomic Landscape of Paediatric Adrenocortical Tumours
ARTICLE Received 5 Aug 2014 | Accepted 16 Jan 2015 | Published 6 Mar 2015 DOI: 10.1038/ncomms7302 Genomic landscape of paediatric adrenocortical tumours Emilia M. Pinto1,*, Xiang Chen2,*, John Easton2, David Finkelstein2, Zhifa Liu3, Stanley Pounds3, Carlos Rodriguez-Galindo4, Troy C. Lund5, Elaine R. Mardis6,7,8, Richard K. Wilson6,7,9, Kristy Boggs2, Donald Yergeau2, Jinjun Cheng2, Heather L. Mulder2, Jayanthi Manne2, Jesse Jenkins10, Maria J. Mastellaro11, Bonald C. Figueiredo12, Michael A. Dyer13, Alberto Pappo14, Jinghui Zhang2, James R. Downing10, Raul C. Ribeiro14,* & Gerard P. Zambetti1,* Paediatric adrenocortical carcinoma is a rare malignancy with poor prognosis. Here we analyse 37 adrenocortical tumours (ACTs) by whole-genome, whole-exome and/or transcriptome sequencing. Most cases (91%) show loss of heterozygosity (LOH) of chromosome 11p, with uniform selection against the maternal chromosome. IGF2 on chromosome 11p is overexpressed in 100% of the tumours. TP53 mutations and chromosome 17 LOH with selection against wild-type TP53 are observed in 28 ACTs (76%). Chromosomes 11p and 17 undergo copy-neutral LOH early during tumorigenesis, suggesting tumour-driver events. Additional genetic alterations include recurrent somatic mutations in ATRX and CTNNB1 and integration of human herpesvirus-6 in chromosome 11p. A dismal outcome is predicted by concomitant TP53 and ATRX mutations and associated genomic abnormalities, including massive structural variations and frequent background mutations. Collectively, these findings demonstrate the nature, timing and potential prognostic significance of key genetic alterations in paediatric ACT and outline a hypothetical model of paediatric adrenocortical tumorigenesis. 1 Department of Biochemistry, St Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA. 2 Department of Computational Biology and Bioinformatics, St Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Genetic Investigation in Non-Obstructive Azoospermia: from the X Chromosome to The
DOTTORATO DI RICERCA IN SCIENZE BIOMEDICHE CICLO XXIX COORDINATORE Prof. Persio dello Sbarba Genetic investigation in non-obstructive azoospermia: from the X chromosome to the whole exome Settore Scientifico Disciplinare MED/13 Dottorando Tutore Dott. Antoni Riera-Escamilla Prof. Csilla Gabriella Krausz _______________________________ _____________________________ (firma) (firma) Coordinatore Prof. Carlo Maria Rotella _______________________________ (firma) Anni 2014/2016 A la meva família Agraïments El resultat d’aquesta tesis és fruit d’un esforç i treball continu però no hauria estat el mateix sense la col·laboració i l’ajuda de molta gent. Segurament em deixaré molta gent a qui donar les gràcies però aquest són els que em venen a la ment ara mateix. En primer lloc voldria agrair a la Dra. Csilla Krausz por haberme acogido con los brazos abiertos desde el primer día que llegué a Florencia, por enseñarme, por su incansable ayuda y por contagiarme de esta pasión por la investigación que nunca se le apaga. Voldria agrair també al Dr. Rafael Oliva per haver fet possible el REPROTRAIN, per haver-me ensenyat moltíssim durant l’època del màster i per acceptar revisar aquesta tesis. Alhora voldria agrair a la Dra. Willy Baarends, thank you for your priceless help and for reviewing this thesis. També voldria donar les gràcies a la Dra. Elisabet Ars per acollir-me al seu laboratori a Barcelona, per ajudar-me sempre que en tot el què li he demanat i fer-me tocar de peus a terra. Ringrazio a tutto il gruppo di Firenze, che dal primo giorno mi sono trovato come a casa. -

Ccessooooooooooooo
THAI MALENG MAT US010017821B2ERIALI MITTE (12 ) United States Patent ( 10 ) Patent No. : US 10 ,017 , 821 B2 Sharp et al. (45 ) Date of Patent : * Jul. 10 , 2018 ( 54 ) BIOMARKERS FOR DIAGNOSING ( 2013 .01 ) ; GOIN 2800 / 324 (2013 . 01 ) ; GOIN ISCHEMIA 2800 / 50 (2013 .01 ) ; GOIN 2800 /7019 ( 2013 .01 ) (71 ) Applicant : The Regents of the University of (58 ) Field of Classification Search California , Oakland , CA (US ) CPC .. .. .. C12Q 1 /6883 ; C12Q 2600 / 158 ; GOIN 33 /6893 ; GOIN 2800 / 2871 ; GOIN ( 72 ) Inventors : Frank Sharp , Davis , CA (US ) ; Glen C 2800 /324 ; GOIN 28 / 50 ; GOIN 2800 /7019 Jickling , Sacramento , CA (US ) See application file for complete search history . ( 73 ) Assignee : The Regents of the University of ( 56 ) References Cited California , Oakland , CA (US ) U . S . PATENT DOCUMENTS ( * ) Notice : Subject to any disclaimer , the term of this 9 ,057 ,109 B2 6 / 2015 Chang patent is extended or adjusted under 35 9 ,200 , 322 B2 12 /2015 Barr et al . U . S . C . 154 ( b ) by 0 days . 9 ,410 ,204 B2 * 8 /2016 Sharp .. .. ... C12Q 1 /6883 This patent is subject to a terminal dis (Continued ) claimer . FOREIGN PATENT DOCUMENTS (21 ) Appl. No. : 15 /226 ,844 WO WO 02 / 12892 2 /2002 WO WO 03 /016910 2 /2003 ( 22 ) Filed : Aug . 2 , 2016 ( Continued ) (65 ) Prior Publication Data OTHER PUBLICATIONS US 2017 /0029891 A1 Feb . 2 , 2017 PCT International Search Report and Written Opinion dated Jul . 25 , Related U . S . Application Data 2008 issued in PCT/ US2008 / 062064 . (63 ) Continuation of application No. 14 /370 ,709 , filed as (Continued ) application No . -

1 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26
1 A Panel of Induced Pluripotent Stem Cells From Chimpanzees: a Resource for Comparative 2 Functional Genomics 3 4 Irene Gallego Romero1,*,†, Bryan J. Pavlovic1,*, Irene Hernando-Herraez2, Xiang Zhou3, Michelle C. 5 Ward1, Nicholas E. Banovich1, Courtney L. Kagan1, Jonathan E. Burnett1, Constance H. Huang1, Amy 6 Mitrano1, Claudia I. Chavarria1, Inbar Friedrich Ben-Nun4,5, Yingchun Li6,7, Karen Sabatini4,7,8 , Trevor 7 R. Leonardo4,7,8, Mana Parast6,7, Tomas Marques-Bonet2,9, Louise C. Laurent7,8, Jeanne F. Loring4, and 8 Yoav Gilad1, † 9 10 1. Department of Human Genetics, University of Chicago, Chicago, IL 60637, USA 11 2. ICREA, Institute of Evolutionary Biology (UPF-CSIC), PRBB, Barcelona 08003, Spain 12 3. Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA 13 4. Center for Regenerative Medicine, Department of Chemical Physiology, The Scripps 14 Research Institute, La Jolla, CA 92037, USA 15 5. Present address: Lonza Walkersville, Inc. 8830 Biggs Ford Road, Walkersville, MD 16 21793, USA 17 6. Department of Pathology, University of California San Diego, San Diego, CA 92103, 18 USA 19 7. Sanford Consortium for Regenerative Medicine, 2880 Torrey Pines Scenic Drive, La 20 Jolla, CA 92037, USA 21 8. Department of Reproductive Medicine, University of California San Diego, San Diego, 22 CA 92103, USA 23 9. Centro Nacional de Análisis Genómico (CNAG), Barcelona, Spain, 08023, Spain 24 25 * These authors contributed equally to this work. 26 † To whom correspondence should be addressed: 27 email: [email protected] 28 email: [email protected] 29 tel: +1 773 702 8507 30 31 Keywords: 32 iPSC, chimpanzee, induced pluripotent stem cells, evolution, Pan troglodytes, cell panel 1 33 Abstract 34 Comparative genomics studies in primates are restricted due to our limited access to samples. -
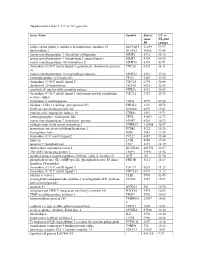
Supplemental Table 2: UC Vs. NL Gene List Gene Name Symbol
Supplemental Table 2: UC vs. NL gene list Gene Name Symbol Entrez UC vs. Gene NL fold ID change solute carrier family 6 (amino acid transporter), member 14 SLC6A14 11254 79.97 dual oxidase 2 DUOX2 50506 73.48 matrix metallopeptidase 1 (interstitial collagenase) MMP1 4312 65.16 matrix metallopeptidase 3 (stromelysin 1, progelatinase) MMP3 4314 60.20 matrix metallopeptidase 10 (stromelysin 2) MMP10 4319 40.71 chemokine (C-X-C motif) ligand 6 (granulocyte chemotactic protein CXCL6 6372 36.11 2) matrix metallopeptidase 12 (macrophage elastase) MMP12 4321 35.94 carboxypeptidase A3 (mast cell) CPA3 1359 33.94 chemokine (C-X-C motif) ligand 5 CXCL5 6374 30.84 cholesterol 25-hydroxylase CH25H 9023 28.08 myeloid cell nuclear differentiation antigen MNDA 4332 24.66 chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating CXCL1 2919 22.55 activity, alpha) tryptophan 2,3-dioxygenase TDO2 6999 21.66 chitinase 3-like 1 (cartilage glycoprotein-39) CHI3L1 1116 20.78 S100 calcium binding protein A8 S100A8 6279 19.81 cysteine-rich, angiogenic inducer, 61 CYR61 3491 19.71 carboxypeptidase, vitellogenic-like CPVL 54504 18.77 matrix metallopeptidase 7 (matrilysin, uterine) MMP7 4316 16.92 collagen triple helix repeat containing 1 CTHRC1 115908 15.47 proprotein convertase subtilisin/kexin type 1 PCSK1 5122 15.26 hemoglobin, beta HBB 3043 13.55 chemokine (C-C motif) ligand 2 CCL2 6347 13.44 lumican LUM 4060 13.43 tenascin C (hexabrachion) TNC 3371 12.19 dual oxidase maturation factor 2 DUOXA2 405753 12.07 TNFAIP3 interacting protein 3 TNIP3 79931 11.96 -

Predicting Human Disease Mutations and Identifying Drug Targets from Mouse Gene Knockout Phenotyping Campaigns Robert Brommage1,*, David R
© 2019. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2019) 12, dmm038224. doi:10.1242/dmm.038224 SPECIAL ARTICLE Predicting human disease mutations and identifying drug targets from mouse gene knockout phenotyping campaigns Robert Brommage1,*, David R. Powell1 and Peter Vogel2 ABSTRACT poorly understood. For example, the Undiagnosed Diseases Two large-scale mouse gene knockout phenotyping campaigns have Network and other DNA sequencing efforts can typically identify provided extensive data on the functions of thousands of mammalian gene mutations for one-third of patients with unknown rare genetic genes. The ongoing International Mouse Phenotyping Consortium diseases (Splinter et al., 2018). The genes, and their actions, (IMPC), with the goal of examining all ∼20,000 mouse genes, responsible for the remaining patients remain unknown. Identifying has examined 5115 genes since 2011, and phenotypic data the actions and biochemical pathways of disease genes provides from several analyses are available on the IMPC website insights for potential therapies. Although imperfect, mice are the (www.mousephenotype.org). Mutant mice having at least one best-established models for human disease (Justice and Dhillon, human genetic disease-associated phenotype are available for 185 2016; Perlman, 2016; Sundberg and Schofield, 2018; Nadeau and IMPC genes. Lexicon Pharmaceuticals’ Genome5000™ campaign Auwerx, 2019). This article summarizes data from two large-scale performed similar analyses between 2000 and the end of 2008 mouse gene knockout phenotyping campaigns: the International focusing on the druggable genome, including enzymes, receptors, Mouse Phenotyping Consortium (IMPC) and Lexicon ’ ™ transporters, channels and secreted proteins. Mutants (4654 genes, Pharmaceuticals Genome5000 program. with 3762 viable adult homozygous lines) with therapeutically Both campaigns employed reverse genetics, the approach that interesting phenotypes were studied extensively. -

Bayesian Infinite Mixture Models for Gene Clustering and Simultaneous
UNIVERSITY OF CINCINNATI Date: 22-Oct-2009 I, Johannes M Freudenberg , hereby submit this original work as part of the requirements for the degree of: Doctor of Philosophy in Biomedical Engineering It is entitled: Bayesian Infinite Mixture Models for Gene Clustering and Simultaneous Context Selection Using High-Throughput Gene Expression Data Student Signature: Johannes M Freudenberg This work and its defense approved by: Committee Chair: Mario Medvedovic, PhD Mario Medvedovic, PhD Bruce Aronow, PhD Bruce Aronow, PhD Michael Wagner, PhD Michael Wagner, PhD Jaroslaw Meller, PhD Jaroslaw Meller, PhD 11/18/2009 248 Bayesian Infinite Mixture Models for Gene Clustering and Simultaneous Context Selection Using High-Throughput Gene Expression Data A dissertation submitted to the Graduate School of the University of Cincinnati in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department Biomedical Engineering of the Colleges of Engineering and Medicine by Johannes M. Freudenberg M.S., 2004, Leipzig University, Leipzig, Germany (German Diplom degree) Committee Chair: Mario Medvedovic, Ph.D. Abstract Applying clustering algorithms to identify groups of co-expressed genes is an important step in the analysis of high-throughput genomics data in order to elucidate affected biological pathways and transcriptional regulatory mechanisms. As these data are becoming ever more abundant the integration with both, existing biological knowledge and other experimental data becomes as crucial as the ability to perform such analysis in a meaningful but virtually unsupervised fashion. Clustering analysis often relies on ad-hoc methods such as k-means or hierarchical clustering with Euclidean distance but model-based methods such as the Bayesian Infinite Mixtures approach have been shown to produce better, more reproducible results.