Mycobacterial Dormancy and Persistence: Molecular Mechanisms Controlling Stress Response, Survival and Adaptation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acetate-Promoted Aldol-Type Reaction: of Acetates and Aldehydes
Acetate PromotedAldol-Condensation Bull. Korean Chem. Soc. 2009, Vol. 30, No. 4 797 Acetate-Promoted Aldol-Type Reaction: Scope and Reactivity of Acetates and Aldehydes Dong Hyeon Kim, A. F. M. Motiur Rahman, Byeong-Seon Jeong, Eung Seok Lee, and Yurngdong Jahng* College of Pharmacy, Yeungnam University, Gyeongsan 712-749, Korea. * E-mail: [email protected] Received November 24, 2008, Accepted February 11, 2009 Potential of acetates and related compounds in glacial acetic acid as a catalyst for aldol-type condensation reactions was examined. Reactions of cycloalkanones or selected heteroaromatics with aldehydes in presence of 10 mol% of various acetates in acetic acid afforded a,a'-bis(substituted-benzylidene)cycloalkanones and substituted-benzyl- idene-mackinazolinones, respectively, in good yields. Among the compounds tested, ammonium acetate is the best and effective especially towards the reactions of mackinazolinone and aliphatic aldehydes to afford 6-alkylidene- mackinazolinones. Key Words: Ammonium acetate, Aldol, 2,5-Bis-benzylidenecyclopentanone, 2,6-Bis-benzylidenecyclo- hexanone, Mackinazolinone Introduction that these were not suitable for a,a'-bis(alkylidene) cyclo- alkanones.21 Previous studies revealed that the reaction of The compounds with a,a'-bis(alkylidene)- and a,a'-bis- cyclopentanone with propionaldehyde in the presence of 0.5 (arylidene)-compounds such as a,a'-bis(benzylidene)cyclo- M NaOH afforded 2,5-bis(ethylidene)cyclopentanone in 19% alkanones (7)1 have been attracting much attention due to not yield21a while the reaction of cyclohexanone with butyralde only their intriguing biological activities such as anti- hyde afforded 3,4-tetramethylene-2-propylbicyclo [3.3.1]no- angiogenic,2 quinine reductase inducer,3 cytotoxic,4 and chole nan-4-ol-9-one (5aa) instead of the expected 2,6-bis(butyl- sterol-lowering activity,5 but also their potentials for nonlinear idene)cyclohexanone (3aa) or 2-butylidenecyclohexanone optical materials.6 They are also the important precursors for (4aa). -

Characterization and Enzyme Engineering of Laccases Towards Lignin Valorization in Aqueous Ionic Liquids
University of Kentucky UKnowledge Theses and Dissertations--Biosystems and Agricultural Engineering Biosystems and Agricultural Engineering 2020 CHARACTERIZATION AND ENZYME ENGINEERING OF LACCASES TOWARDS LIGNIN VALORIZATION IN AQUEOUS IONIC LIQUIDS Joseph Stevens University of Kentucky, [email protected] Author ORCID Identifier: https://orcid.org/0000-0002-2595-9256 Digital Object Identifier: https://doi.org/10.13023/etd.2020.346 Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Stevens, Joseph, "CHARACTERIZATION AND ENZYME ENGINEERING OF LACCASES TOWARDS LIGNIN VALORIZATION IN AQUEOUS IONIC LIQUIDS" (2020). Theses and Dissertations--Biosystems and Agricultural Engineering. 72. https://uknowledge.uky.edu/bae_etds/72 This Doctoral Dissertation is brought to you for free and open access by the Biosystems and Agricultural Engineering at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Biosystems and Agricultural Engineering by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. -

Riedel-De Haentm
Riedel-deRdH HaenTM z BBurdickJa + HoneywellHw = PProroductivity 2 & JacksonTM FlkFlukaTM Discover the perfect formula EMEA, Russia & CIS Product Catalog Trusted Supplier of Chemicals and Reagents At Honeywell, we know that working on the cutting edge is never simple. To be successful you need a range of consistent high-quality products, access to flexible solutions, and expert support. That’s why we are committed to providing more high-quality products, custom capabilities and technical help that meet our customers’ requirements. Consistent, High Quality Products Over 200 years of experience and innovation in producing high quality solvents and reagents enables us to reliably manufacture consistent products for every lot we produce. The vast majority of our products are produced in Seelze, Germany or Muskegon, Michigan, USA. Both sites are certified to meet ISO 9001 and ISO 14001 which helps us guarantee the quality and consistency of our products. Customization and Choice We offer a comprehensive and growing range of solvents and reagents, from industry leading brands such as Honeywell Fluka™, Honeywell Support When You Need It Burdick & Jackson™, and Honeywell Riedel-de We take product and customer support Haën™, suitable for use in a broad range of seriously. All of our customer service applications. With our new state-of-the-art representatives are trained to be responsive, distribution centers and extensive network of helpful and knowledgeable about our products. distributor partners, we can now deliver our Whether you want to place an order, track a portfolio to almost anywhere in the world. delivery or simply contact us to ask a question, Because of our global footprint, we are well you will receive a courteous and accurate positioned to listen to our customers and response. -

Nucleoside Analogues for Positron Emission Tomography Imaging and to Study Radiation Mediated Generation of Radicals from Azides
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 6-27-2019 Nucleoside Analogues for Positron Emission Tomography Imaging and to Study Radiation Mediated Generation of Radicals from Azides Maria E. de Cabrera Florida International University, [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Nucleic Acids, Nucleotides, and Nucleosides Commons Recommended Citation de Cabrera, Maria E., "Nucleoside Analogues for Positron Emission Tomography Imaging and to Study Radiation Mediated Generation of Radicals from Azides" (2019). FIU Electronic Theses and Dissertations. 4229. https://digitalcommons.fiu.edu/etd/4229 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida NUCLEOSIDE ANALOGUES FOR POSITRON EMISSION TOMOGRAPHY IMAGING AND TO STUDY RADIATION MEDIATED GENERATION OF RADICALS FROM AZIDES A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR IN PHILOSOPHY in CHEMISTRY by Maria de Cabrera 2019 To: Dean Michael R. Heithaus College of Arts, Sciences and Education This dissertation, written by Maria de Cabrera, and entitled Nucleoside Analogues for Positron Emission Tomography Imaging and to Study Radiation Mediated Generation of Radicals from Azides, having been approved in respect to style and intellectual content, is referred to you for judgment. We have read this dissertation and recommend that it be approved. __________________________________________ Raphael Raptis __________________________________________ Kathleen Rein __________________________________________ Jose Almirall __________________________________________ M. -

United States Patent (19) (11) Patent Number: 5,002,885 Stavrianopoulos (45) Date of Patent: Mar
United States Patent (19) (11) Patent Number: 5,002,885 Stavrianopoulos (45) Date of Patent: Mar. 26, 1991 (54) DETECTABLE MOLECULES, METHOD oyl, carbonyl, carboxy, thiol or a residue comprising an PREPARATION AND USE activated carbon; -X- is selected from the group 75 Inventor: Jannis G. Stavrianopoulos, New consisting of York, N.Y. O NH 73) Assignee: Enzo Biochem, Inc., New York, N.Y. -NH-C-, -NH-e Cas, -N=N-ter, 21 Appl. No.: 513,723 -NHSO-, as OSO-e, -NH-N=N-, (22 Filed: Apr. 24, 1990 O O Related U.S. Application Data -NHale C-CH-e NH-, -ao CH-NH-, -O-C-, 60 Continuation of Ser. No. 43,668, Apr. 28, 1987, Pat. No. 4,952,685, Division of Ser. No. 575,396, Jan. 30, 1984, Pat. No. 4,707,440. -NH-C-CH2-es-, -NH-CH2-, -e-o-c-CH2, 51 Int. Cl. ....................... C12N 9/96; CO7K 15/00; CO7K 15/28 52 U.S. Cl. ........................................ 435/188; 435/6; 435/7.5; 435/7.9; 435/7.92; 436/548; 530/350; 530/387; 530/339; 530/390; 530/391; 530/402; 536/1.1; 536/55.1; 536/56; 536/102; 536/27; -R1- is 536/28; 536/29 58 Field of Search ............... 530/350, 402, 387, 389, 530/390, 391; 435/188, 7, 6; 536/27-29, 1.1, 55.1, 56, 102; 436/548 a C1-C10 branched or unbranched alkyl or aralkyl, 56 References Cited which may be substituted by -OH, -Y- is a direct U.S. PATENT DOCUMENTS bond to -E-, or -Y- is -E-R2- where R2 is a 4,298,685 11/1981 Parikh et al. -
Biomarkers of Exposure to Nitrosation Products of Amino Acids and Peptides
Biomarkers of exposure to nitrosation products of amino acids and peptides. Thesis submitted for the degree of Doctor of Philosophy at the University of Leicester by Kathryn Lisa Harrison BSc. MSc. CMHT University of Leicester July 1998 UMI Number: U536777 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. Dissertation Publishing UMI U536777 Published by ProQuest LLC 2013. Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract Biomarkers of exposure to nitrosation products of amino acids and peptides. Kathryn L. Harrison. The endogenous nitrosation of dietary amino acids and peptides may be a major source of genotoxic damage in the stomach and lower GI tract. Amongst the many N-alkyl-N- nitrosocompounds and related compounds, there are a number which share the common features of being derived from glycine and are carboxymethylating agents. Novel immunoaffinity-RP-HPLC-fluorescence methods were developed allowing the concomitant detection of 0 6-carboxymethy 1-2 ’ -deoxyguanosine (0 6-CMdG) and 0 6-methyl- 2’-deoxyguanosine (0 6-MedG) with limits of detection of 0.128pmol 0 6-CMdG/mol dG and 0.064(j.mol 0 6-MedG/mol dG in lmg of DNA. -

An Invitation for Bid (IFB) #1207170535: Disposal of Chemicals and Chemical Waste
NEW YORK STATE DEPARTMENT OF HEALTH An Invitation for Bid (IFB) for Disposal of Chemicals and Chemical Waste Wadsworth Center Safety and Security Office FAU#1207170535 Completed Questions Below are written questions and questions received at the pre-bid conference or within the open Q&A period, and the corresponding responses. Question 1 Q: It is difficult to estimate costs per container based on the information provided [in the bid package]. Should an average estimate be used? Can the New York State Department of Health (NYS DOH) Wadsworth Center (generator) provide a list of drum sizes/contents for the previous year based on the manifests? A: Yes. Please see attached Q&A Table A for a list that includes the Department of Transportation (DOT) description, quantity, container size (in gallons), and EPA codes. Question 2 Q: For category A1 [on the bid sheet], can the NYS DOH Wadsworth Center (generator) provide an inventory list? A: Yes. Please see attached Q&A Table B listing waste collected by the NYS DOH Wadsworth Center (generator) for the past 2½ years. Question 3 Q: Can the NYS DOH Wadsworth Center (generator) provide profiles of bulk solvent chemicals as well? A: No. The NYS DOH Wadsworth Center (generator) cannot provide copies of the actual profiles. However, bulk materials are of the following general categories: Oils, lab pack (code: LABP), waste solvent (code: D001, examples of waste include: acetonitrile/methanol and formalin/methanol, xylene, hexane, alcohol, and dichloromethane waste), and waste guanidine thiocyanate buffer -

Ep001402024b1*
(19) *EP001402024B1* (11) EP 1 402 024 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12P 1/00 (2006.01) 22.08.2007 Bulletin 2007/34 C12P 19/34 (2006.01) C07H 21/00 (2006.01) C12P 21/02 (2006.01) (21) Application number: 02740409.4 (86) International application number: (22) Date of filing: 20.06.2002 PCT/DK2002/000419 (87) International publication number: WO 2002/103008 (27.12.2002 Gazette 2002/52) (54) TEMPLATED MOLECULES AND METHODS FOR USING SUCH MOLECULES EINE METHODE ZUR SYNTHESE VON MATRIZENABHÄNGIGEN MOLEKÜLEN MOLECULES A MATRICE ET PROCEDES D’UTILISATION DE CES DERNIERES (84) Designated Contracting States: • FRANCH, Thomas AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU DK-5000 Odense C (DK) MC NL PT SE TR • THISTED, Thomas Designated Extension States: DK-3600 Frederikssund (DK) AL LT LV MK RO SI • HYLDTOFT, Lene DK-2830 Virum (DK) (30) Priority: 20.06.2001 DK 200100962 • NØRREGAARD-MADSEN, Mads 15.03.2002 DK 200200415 DK-3460 Birkerød (DK) • ANDERS GODSKESEN, Michael (43) Date of publication of application: DK-2950 Vedbæk (DK) 31.03.2004 Bulletin 2004/14 • SCHRØDER GLAD, Sanne DK-2750 Ballerup (DK) (73) Proprietor: Nuevolution A/S 2100 Copenhagen (DK) (74) Representative: HOEIBERG A/S European Patent & Trademark Attorneys (72) Inventors: Store Kongensgade 59A • PEDERSEN, Henrik 1264 Copenhagen K (DK) DK-2880 Bagsværd (DK) • GOUILAEV, Alex, Haahr (56) References cited: 3670 Veksø Sjælland (DK) WO-A-00/23458 WO-A-00/61775 • SAMS, Klarner, Christian WO-A-93/03172 WO-A-98/56904 DK-1973 Frederiksberg C (DK) WO-A-02/074929 DE-C- 19 646 372 • SLØK, Frank, Abilgaard DK-2200 København N (DK) • WALDER J A ET AL: "COMPLEMENTARY • FRESKGÅRD, PER-OLA CARRIER PEPTIDE SYNTHESIS: GENERAL DK-235 93 Vellinge (SE) STRATEGY AND IMPLICATIONS FOR • HOLTMANN, Anette PREBIOTIC ORIGIN OF PEPTIDE SYNTHESIS" DK-2750 Ballerup (DK) PROCEEDINGS OF THE NATIONAL ACADEMY • KAMPMANN OLSEN, Eva OF SCIENCES OF USA, NATIONAL ACADEMY OF DK-2730 Herlev (DK) SCIENCE. -

Process for Preparing Fluorine-Containing Dicarbonyl
Europäisches Patentamt *EP000781752B1* (19) European Patent Office Office européen des brevets (11) EP 0 781 752 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.7: C07C 45/63, C07B 39/00 of the grant of the patent: 21.02.2001 Bulletin 2001/08 (21) Application number: 97103636.3 (22) Date of filing: 01.11.1993 (54) Process for preparing fluorine-containing dicarbonyl compound Verfahren zur Herstellung von fluorierten Dicarbonylverbindungen Procédé de préparation de composés dicarbonylés fluorés (84) Designated Contracting States: • Tomizawa, Ginjiro, MEC Kenkyusho CH DE FR GB IT LI Tsukuba-shi, Ibaraki 305 (JP) (30) Priority: 30.10.1992 JP 31580092 (74) Representative: Hansen, Bernd, Dr. Dipl.-Chem. et al (43) Date of publication of application: Hoffmann Eitle, 02.07.1997 Bulletin 1997/27 Patent- und Rechtsanwälte, Arabellastrasse 4 (62) Document number(s) of the earlier application(s) in 81925 München (DE) accordance with Art. 76 EPC: 93923678.2 / 0 667 332 (56) References cited: • JOURNAL OF ORGANIC CHEMISTRY, vol. 52, (73) Proprietor: DAIKIN INDUSTRIES, LIMITED no. 19, 1987, EASTON US, pages 4307-4310, Osaka-shi Osaka 530 (JP) XP002029446 SUZANNE T.PURRINGTON ET AL: "Selective monofluorination of beta-diketones" (72) Inventors: • Umemoto, Teruo, MEC Kenkyusho Tsukuba-shi, Ibaraki 305 (JP) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. Notice of opposition shall be filed in a written reasoned statement. It shall not be deemed to have been filed until the opposition fee has been paid. -
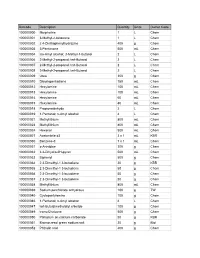
Web Inventory Aug 08
Barcode Description Quantity Units Owner Code 100000000 Morpholine 1 L Chem 100000001 3-Methyl-2-butanone 1 L Chem 100000002 2 4-Dinitrophenylhydrazine 400 g Chem 100000003 3-Pentanone 500 mL Chem 100000004 iso-Amyl alcohol; 3-Methyl-1-butanol 2 L Chem 100000006 2-Methyl-2-propanol; tert-Butanol 2 L Chem 100000007 2-Methyl-2-propanol; tert-Butanol 2 L Chem 100000008 2-Methyl-2-propanol; tert-Butanol 2 L Chem 100000009 Urea 300 g Chem 100000010 Dicyclopentadiene 750 mL Chem 100000012 Hexylamine 100 mL Chem 100000013 Hexylamine 100 mL Chem 100000014 Hexylamine 60 mL Chem 100000015 Hexylamine 80 mL Chem 100000018 Propionaldehyde 2 L Chem 100000019 1-Pentanol; n-Amyl alcohol 4 L Chem 100000021 Methyllithium 800 mL Chem 100000023 Methyllithium 800 mL Chem 100000024 Hexanal 500 mL Chem 100000027 Acetonitrile d3 3 x 1 mL KBR 100000030 Benzene-d 7 x 1 mL Chem 100000031 o-Anisidine 300 g Chem 100000032 3 4-Dihydro-2H-pyran 500 mL Chem 100000033 Biphenyl 500 g Chem 100000034 2 3-Dimethyl-1 3-butadiene 20 g KBR 100000035 2 3-Dimethyl-1 3-butadiene 50 g Chem 100000036 2 3-Dimethyl-1 3-butadiene 50 g Chem 100000037 2 3-Dimethyl-1 3-butadiene 50 g Chem 100000038 Methyllithium 800 mL Chem 100000039 Sodium perchlorate anhydrous 100 g TW 100000040 Cyclopentylamine 100 g Chem 100000046 1-Pentanol; n-Amyl alcohol 4 L Chem 100000047 tert-Butyldimethylsilyl chloride 100 g Chem 100000049 trans-Chalcone 500 g Chem 100000050 Palladium on calcium carbonate 50 g KBR 100000051 Bromocresol green sodium salt 20 g Biol 100000052 Phthalic acid 400 g Chem 100000053 4-Heptanone -

Process for Preparing Fluorine-Containing Dicarbonyl Compound
* — mi mi ii mi ii i iii 1 1 ii ii i ii OJII Eur°Pean Patent Office <*S Office europeen des brevets (11) EP 0 781 752 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) int. CI.6: C07C 45/63, C07B 39/00 02.07.1997 Bulletin 1997/27 (21) Application number: 97103636.3 (22) Date of filing: 01.11.1993 (84) Designated Contracting States: • Tomizawa, Ginjiro, CH DE FR GB IT LI MEC Kenkyusho Tsukuba-shi, Ibaraki 305 (JP) (30) Priority: 30.10.1992 JP 315800/92 (74) Representative: Hansen, Bernd, Dr. Dipl.-Chem. (62) Document number(s) of the earlier application(s) in et al accordance with Art. 76 EPC: Hoffmann, Eitle & Partner, 93923678.2 / 0 667 332 Patentanwalte, ArdbolldstrdssG 4 (71) Applicant: DAI KIN INDUSTRIES, LIMITED 81925 Munchen (DE) Osaka-shi Osaka 530 (JP) Remarks: (72) Inventors: This application was filed on 05 - 03 - 1 997 as a • Umemoto, Teruo, divisional application to the application mentioned MEC Kenkyusho under INID code 62. Tsukuba-shi, Ibaraki 305 (JP) (54) Process for preparing fluorine-containing dicarbonyl compound (57) A process for preparing a fluorine-containing dicarbonyl compound of the formula: R1COCFR2COR3 in which R1 is a hydrogen atom, or a substituted or unsubstituted alkyl or aryl group; R2 is a hydrogen atom, a halogen atom, or a substituted or unsubstituted alkyl or aryl group; and R3 is a hydrogen atom, or a substi- tuted or unsubstituted alkyl, aryl, alkoxy or aryloxy group, provided that at least two of R1 , R2 and R3 may together form a part of a cyclic structure with or without a hetero atom, by reacting a dicarbonyl compound of the formula: R1COCHR2COR3 in which R1, R2 and R3 are the same as defined above with fluorine (F2) in a solvent in the presence of a salt, or an acid having pKa of 6 or less. -

United States Patent (19) 11 Patent Number: 4,849,505 Stavrianopoulos 45 Date of Patent: Jul
United States Patent (19) 11 Patent Number: 4,849,505 Stavrianopoulos 45 Date of Patent: Jul. 18, 1989 (54) DETECTABLEMOLECULES, METHOD OF activated carbon; -X- is selected from the group PREPARATION AND USE consisting of 75 Inventor: Jannis G. Stavrianopoulos, New York, N.Y. 73) Assignee: Enzo Biochem, Inc., New York, N.Y. 21 Appl. No.: 43,572 (22 Filed: Apr. 28, 1987 Related U.S. Application Data O O 62 Division of Ser. No. 575,396, Jan. 30, 1984, Pat. No. 4,707,440. O l 51 Int. Cl." ...................... C07H 15/12; C07K 15/00 -O-C-CH2, -S-CH2-, -O-CO-NH 52 U.S. Cl. ..................................... 530/300; 424/1.1; 435/5; 435/6; 435/7; 435/180; 436/518; -R1- is 436/531; 436/532; 530/350, 530/402; 530/405; 536/26; 536/27; 536/55.1 58 Field of Search .................... 424/1.1; 435/5, 6, 7, 435/180; 436/518, 531,532; 530/300, 350, 402, 405; 536/26, 27, 55.1 (56) References Cited U.S. PATENT DOCUMENTS 4,298,685 11/1981 Parikh et al. ........................... 435/7 4,374,120 2/1983 Soini et al. ...... ... 436/546 or a C1-C10 branched or unbranched alkyl or aralkyl, 4,472,509 9/1984 Gansow et al. ..................... 436/548 which may be substituted by -OH, -Y- is a direct 4,535,057 8/1985 Dreesman et al. ...................... 435/5 bond to -E-, or -Y- is -E-R2- where R2 is a 4,563,419 1/1986 Ranki et al. ........................... 435/9 C1-C10 branched or unbranched alkyl; Za is chlorine, 4,637,988 1/1987 Hinshaw et al.