PDF (Ashbythesis.Pdf)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Assembly of the Cnidarian Camera-Type Eye from Vertebrate-Like Components
Assembly of the cnidarian camera-type eye from vertebrate-like components Zbynek Kozmik*†, Jana Ruzickova*‡, Kristyna Jonasova*‡, Yoshifumi Matsumoto§, Pavel Vopalensky*, Iryna Kozmikova*, Hynek Strnad*, Shoji Kawamura§, Joram Piatigorsky†¶, Vaclav Paces*, and Cestmir Vlcek*† *Institute of Molecular Genetics, Academy of Sciences of the Czech Republic, Videnska 1083, 142 20 Prague 4, Czech Republic; §Graduate School of Frontier Sciences, University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8562, Japan; and ¶National Eye Institute, National Institutes of Health, Bethesda, MD 20892-3655 Edited by Eviatar Nevo, University of Haifa, Haifa, Israel, and approved April 11, 2008 (received for review January 14, 2008) Animal eyes are morphologically diverse. Their assembly, however, containing Tripedalia eyes have sophisticated visual optics as do always relies on the same basic principle, i.e., photoreceptors advanced bilaterian phyla (11). located in the vicinity of dark shielding pigment. Cnidaria as the In the present work, we characterize genes required for the likely sister group to the Bilateria are the earliest branching phylum assembly of camera-type eyes in Tripedalia. We show that the with a well developed visual system. Here, we show that camera- genetic building blocks typical of vertebrate eyes, namely ciliary type eyes of the cubozoan jellyfish, Tripedalia cystophora, use opsin and the melanogenic pathway, are used by the cubozoan genetic building blocks typical of vertebrate eyes, namely, a ciliary eyes. Although our findings of unsuspected parallelism are phototransduction cascade and melanogenic pathway. Our find- consistent with either an independent origin or common ances- ings indicative of parallelism provide an insight into eye evolution. try of cubozoan and vertebrate eyes, we believe the present data Combined, the available data favor the possibility that vertebrate favor the former alternative. -

1 Eric Davidson and Deep Time Douglas H. Erwin Department Of
Eric Davidson and Deep Time Douglas H. Erwin Department of Paleobiology, MRC-121 National Museum of Natural History Washington, DC 20013-7012 E-mail: [email protected] Abstract Eric Davidson had a deep and abiding interest in the role developmental mechanisms played in the generating evolutionary patterns documented in deep time, from the origin of the euechinoids to the processes responsible for the morphological architectures of major animal clades. Although not an evolutionary biologist, Davidson’s interests long preceded the current excitement over comparative evolutionary developmental biology. Here I discuss three aspects at the intersection between his research and evolutionary patterns in deep time: First, understanding the mechanisms of body plan formation, particularly those associated with the early diversification of major metazoan clades. Second, a critique of early claims about ancestral metazoans based on the discoveries of highly conserved genes across bilaterian animals. Third, Davidson’s own involvement in paleontology through a collaborative study of the fossil embryos from the Ediacaran Doushantuo Formation in south China. Keywords Eric Davidson – Evolution – Gene regulatory networks – Bodyplan – Cambrian Radiation – Echinoderms 1 Introduction Eric Davidson was a developmental biologist, not an evolutionary biologist or paleobiologist. He was driven to understand the mechanisms of gene regulatory control and how they controlled development, but this focus was deeply embedded within concerns about the relationship between development and evolution. Questions about the origin of major metazoan architectures or body plans were central to Eric’s concerns since at least the late 1960s. His 1971 paper with Roy Britten includes a section on “The Evolutionary Growth of the Genome” illustrated with a figure depicting variations in genome size in major animal groups and a metazoan phylogeny (Britten and Davidson 1971). -

Systems Biology Lecture Outline (Systems Biology) Michael K
Spring 2019 – Epigenetics and Systems Biology Lecture Outline (Systems Biology) Michael K. Skinner – Biol 476/576 CUE 418, 10:35-11:50 am, Tuesdays & Thursdays January 22 & 29, 2019 Weeks 3 and 4 Systems Biology (Components & Technology) Components (DNA, Expression, Cellular, Organ, Physiology, Organism, Differentiation, Development, Phenotype, Evolution) Technology (Genomics, Transcriptomes, Proteomics) (Interaction, Signaling, Metabolism) Omics (Data Processing and Resources) Required Reading ENCODE (2012) ENCODE Explained. Nature 489:52-55. Tavassoly I, Goldfarb J, Iyengar R. (2018) Essays Biochem. 62(4):487-500. Literature Sundaram V, Wang T. Transposable Element Mediated Innovation in Gene Regulatory Landscapes of Cells: Re-Visiting the "Gene-Battery" Model. Bioessays. 2018 40(1). doi: 10.1002/bies.201700155. Abil Z, Ellefson JW, Gollihar JD, Watkins E, Ellington AD. Compartmentalized partnered replication for the directed evolution of genetic parts and circuits. Nat Protoc. 2017 Dec;12(12):2493- 2512. Filipp FV. Crosstalk between epigenetics and metabolism-Yin and Yang of histone demethylases and methyltransferases in cancer. Brief Funct Genomics. 2017 Nov 1;16(6):320-325. Chen Z, Li S, Subramaniam S, Shyy JY, Chien S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu Rev Biomed Eng. 2017 Jun 21;19:195-219. Hollick JB. Paramutation and related phenomena in diverse species. Nat Rev Genet. 2017 Jan;18(1):5-23. Macovei A, Pagano A, Leonetti P, Carbonera D, Balestrazzi A, Araújo SS. Systems biology and genome-wide approaches to unveil the molecular players involved in the pre-germinative metabolism: implications on seed technology traits. Plant Cell Rep. 2017 May;36(5):669-688. Bagchi DN, Iyer VR. -

The Human Proteome Co-Regulation Map Reveals Functional Relationships Between Proteins
bioRxiv preprint doi: https://doi.org/10.1101/582247; this version posted March 19, 2019. The copyright holder for this preprint (which was 19/03/2019not certified by peer review) is the author/funder, who hasNBT_manuscript_revised granted bioRxiv a license - toGoogle display Docs the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 The human proteome co-regulation map reveals functional relationships 2 between proteins 3 Georg Kustatscher1 *, Piotr Grabowski2 *, Tina A. Schrader3 , Josiah B. Passmore3 , Michael 4 Schrader3 , Juri Rappsilber1,2# 5 1 Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh EH9 3BF, UK 6 2 Bioanalytics, Institute of Biotechnology, Technische Universität Berlin, 13355 Berlin, 7 Germany 8 3 Biosciences, University of Exeter, Exeter EX4 4QD, UK 9 * Equal contribution 10 # Communicating author: [email protected] 11 Submission as Resource article. 12 The annotation of protein function is a longstanding challenge of cell biology that 13 suffers from the sheer magnitude of the task. Here we present ProteomeHD, which 14 documents the response of 10,323 human proteins to 294 biological perturbations, 15 measured by isotope-labelling mass spectrometry. Using this data matrix and robust 16 machine learning we create a co-regulation map of the cell that reflects functional 17 associations between human proteins. The map identifies a functional context for 18 many uncharacterized proteins, including microproteins that are difficult to study with 19 traditional methods. Co-regulation also captures relationships between proteins 20 which do not physically interact or co-localize. For example, co-regulation of the 21 peroxisomal membrane protein PEX11β with mitochondrial respiration factors led us 22 to discover a novel organelle interface between peroxisomes and mitochondria in 23 mammalian cells. -

Xenacoelomorpha's Significance for Understanding Bilaterian Evolution
Available online at www.sciencedirect.com ScienceDirect Xenacoelomorpha’s significance for understanding bilaterian evolution Andreas Hejnol and Kevin Pang The Xenacoelomorpha, with its phylogenetic position as sister biology models are the fruitfly Drosophila melanogaster and group of the Nephrozoa (Protostomia + Deuterostomia), plays the nematode Caenorhabditis elegans, in which basic prin- a key-role in understanding the evolution of bilaterian cell types ciples of developmental processes have been studied in and organ systems. Current studies of the morphological and great detail. It might be because the field of evolutionary developmental diversity of this group allow us to trace the developmental biology — EvoDevo — has its origin in evolution of different organ systems within the group and to developmental biology and not evolutionary biology that reconstruct characters of the most recent common ancestor of species under investigation are often called ‘model spe- Xenacoelomorpha. The disparity of the clade shows that there cies’. Criteria for selected representative species are cannot be a single xenacoelomorph ‘model’ species and primarily the ease of access to collected material and strategic sampling is essential for understanding the evolution their ability to be cultivated in the lab [1]. In some cases, of major traits. With this strategy, fundamental insights into the a supposedly larger number of ancestral characters or a evolution of molecular mechanisms and their role in shaping dominant role in ecosystems have played an additional animal organ systems can be expected in the near future. role in selecting model species. These arguments were Address used to attract sufficient funding for genome sequencing Sars International Centre for Marine Molecular Biology, University of and developmental studies that are cost-intensive inves- Bergen, Thormøhlensgate 55, 5008 Bergen, Norway tigations. -

Molecular Homology & the Ancient Genetic Toolkit: How Evolutionary
Regis University ePublications at Regis University All Regis University Theses Spring 2019 Molecular Homology & the Ancient Genetic Toolkit: How Evolutionary Development Could Shape Your Next Doctor's Appointment Elizabeth G. Plender Follow this and additional works at: https://epublications.regis.edu/theses Part of the Cell Biology Commons, Developmental Biology Commons, Evolution Commons, Molecular Biology Commons, and the Molecular Genetics Commons Recommended Citation Plender, Elizabeth G., "Molecular Homology & the Ancient Genetic Toolkit: How Evolutionary Development Could Shape Your Next Doctor's Appointment" (2019). All Regis University Theses. 905. https://epublications.regis.edu/theses/905 This Thesis - Open Access is brought to you for free and open access by ePublications at Regis University. It has been accepted for inclusion in All Regis University Theses by an authorized administrator of ePublications at Regis University. For more information, please contact [email protected]. MOLECULAR HOMOLOGY AND THE ANCIENT GENETIC TOOLKIT: HOW EVOLUTIONARY DEVELOPMENT COULD SHAPE YOUR NEXT DOCTOR’S APPOINTMENT A thesis submitted to Regis College The Honors Program In partial fulfillment of the requirements For Graduation with Honors By Elizabeth G. Plender May 2019 Thesis written by Elizabeth Plender Approved by Thesis Advisor Thesis Reader Accepted by Director, University Honors Program ii iii TABLE OF CONTENTS List of Figures ............................................................................................................................................... -
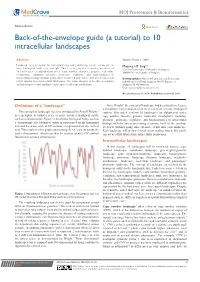
Back-Of-The-Envelope Guide (A Tutorial) to 10 Intracellular Landscapes
MOJ Proteomics & Bioinformatics Review Article Open Access Back-of-the-envelope guide (a tutorial) to 10 intracellular landscapes Abstract Volume 7 Issue 1 - 2018 Landscape is a metaphor for conceptualizing and visualizing a score across one or Maurice HT Ling1,2 more biological entities or concepts. This review provides a cursory overview of 1Colossus Technologies LLP, Republic of Singapore 10 landscapes (in alphabetical order, copy number, fluxome, genome, molecular, 2HOHY Pte. Ltd., Republic of Singapore metabolome, mutation, phenome, proteome, regulome, and transcriptome) in intracellular biology without going into extensive depth; hence, this article can act as Correspondence: Maurice HT Ling, Colossus Technologies a first tutorial into intracellular landscapes. The value ahead is to be able to compare LLP, 8 Burns Road, Trivex, Singapore 369977, Republic of and interrogate across multiple landscapes at different resolutions. Singapore, Tel +6596669233, Email [email protected] Received: January 29, 2018 | Published: February 06, 2018 Definition of a “landscape” Since Wright,1 the concept of landscape had been used to reference a metaphoric representation of an overview of one or more biological 1 The concept of landscape was first introduced by Sewell Wright, entities. This article reviews 10 landscapes (in alphabetical order, as a metaphor to visual a scale or score across a biological entity, copy number, fluxome, genome, molecular, metabolome, mutation, such as a chromosome (Figure 1). In a linear biological entity, such as phenome, proteome, regulome, and transcriptome) in intracellular a chromosome; the biological entity is represented on the horizontal biology with the aim of presenting a cursory, back of the envelope axis and the score, such as GC content, is represented on the vertical overview without going into extensive depth into each landscape. -

The Palaeoscolecida and the Evolution of the Ecdysozoa Andrey Yu
The Palaeoscolecida and the evolution of the Ecdysozoa Andrey Yu. Zhuravlev1, José Antonio Gámez Vintaned2 and Eladio Liñán1 1Área y Museo de Paleontología, Departamento de Ciencias de la Tierra, Facultad de Ciencias, Universidad de Zaragoza, C/ Pedro Cerbuna, 12, E-50009 Zaragoza, Spain 2Área de Paleontología, Departamento de Geologica, Facultad de Ciencias Biológicas, Univeristat de València, C/ Doctor Moliner, 50, E-46100 Burjassot, Spain Email: [email protected] AbstrAct rÉsUMÉ Palaeoscolecidans are a key group for understanding the ear- Les Paléoscolécides sont un groupe clé pour la compréhen- ly evolution of the Ecdysozoa. The Palaeoscolecida possess sion des débuts de l’évolution des Ecdysozoa. Les Pal- a terminal mouth and an anus, an invertible proboscis with aeoscolecida possèdent une bouche terminale et un anus, pointed scalids, a thick integument of diverse plates, sensory un proboscis inversible aux scalides pointues, un tégument papillae and caudal hooks. These are features that draw a épais de plaques diverses, des papilles sensorielles et des secret out of these worms, indicating palaeoscolecidan af- crochets caudaux. Ceux-ci sont des traits qui tirent un secret finities with the phylum Cephalorhyncha, which embraces de ces vers, ce qui indique des affinités paléoscolecides avec priapulids, kinorhynchs, loriciferans and nematomorphs. At le phylum des Cephalorhyncha qui inclut les priapulides, les the same time, the Palaeoscolecida share a number of char- kinorhynches, les loricifères et les nématomorphes. Cepen- acters with the lobopod-bearing Cambrian ecdysozoans, the dant, les Palaeoscolecida ont aussi quelques-uns des mêmes Xenusia. Xenusians commonly possess a terminal mouth, caractères que les Xenusia, ces écdysozaires cambriens qui a proboscis (although not retractable), and a thick integu- portaient des lobopodes. -

Bioinformatics Strategies for Identification of Cancer Biomarkers and Targets in Pathogens Associated with Cancer
FEDERAL UNIVERSITY OF MINAS GERAIS INSTITUTE OF BIOLOGICAL SCIENCE DEPARTMENT OF GENERAL BIOLOGY GRADUATE PROGRAM OF BIOINFORMATICS BIOINFORMATICS STRATEGIES FOR IDENTIFICATION OF CANCER BIOMARKERS AND TARGETS IN PATHOGENS ASSOCIATED WITH CANCER PhD STUDENT: Debmalya Barh SUPERVISOR: Prof. Dr. Vasco Ariston de Carvalho Azevedo Page 1 of 237 DEBMALYA BARH Bioinformatics strategies for identification of cancer biomarkers and targets in pathogens associated with cancer PhD STUDENT: Debmalya Barh SUPERVISOR: Prof. Dr. Vasco Ariston de Carvalho Azevedo FEDERAL UNIVERSITY OF MINAS GERAIS INSTITUTE OF BIOLOGICAL SCIENCES BELO HORIZONTE - MG February – 2017 Page 2 of 237 043 Barh, Debmalya. Bioinformatics strategies for identification of cancer biomarkers and targets in pathogens associated with cancer [manuscrito] / Debmalya Barh. – 2017. 237 f. : il. ; 29,5 cm. Supervisor: Vasco Ariston de Carvalho Azevedo. Tese (doutorado) – Federal University of Minas Gerais, Institute of Biological Sciences. 1. Bioinformática - Teses. 2. Marcadores biológicos. 3. Doenças transmissíveis - Teses. 4. Transcriptômica - Teses. 5. MicroRNAs - Teses. I. Azevedo , Vasco Ariston de Carvalho. II. Universidade Federal de Minas Gerais. Instituto de Ciências Biológicas. III. Título. CDU: 573:004 TABLE OF CONTENTS TABLE OF CONTENTS……………………………………………………………………………………i DEDICATION………………………………………………………………………………………..……iii ACKNOWLEDGEMENTS…………………………………………………………………………..……iv LIST OF ABBREVIATIONS…………………………………………………………………….……….. v ABSTRACT………………………………………………………………………………………...……..vii -

Can Molecular Clocks and the Fossil Record Be Reconciled?
Prospects & Overviews Review essays The origin of animals: Can molecular clocks and the fossil record be reconciled? John A. Cunningham1)2)Ã, Alexander G. Liu1)†, Stefan Bengtson2) and Philip C. J. Donoghue1) The evolutionary emergence of animals is one of the most Introduction significant episodes in the history of life, but its timing remains poorly constrained. Molecular clocks estimate that The apparent absence of a fossil record prior to the appearance of trilobites in the Cambrian famously troubled Darwin. He animals originated and began diversifying over 100 million wrote in On the origin of species that if his theory of evolution years before the first definitive metazoan fossil evidence in were true “it is indisputable that before the lowest [Cambrian] the Cambrian. However, closer inspection reveals that clock stratum was deposited ... the world swarmed with living estimates and the fossil record are less divergent than is creatures.” Furthermore, he could give “no satisfactory answer” often claimed. Modern clock analyses do not predict the as to why older fossiliferous deposits had not been found [1]. In the intervening century and a half, a record of Precambrian presence of the crown-representatives of most animal phyla fossils has been discovered extending back over three billion in the Neoproterozoic. Furthermore, despite challenges years (popularly summarized in [2]). Nevertheless, “Darwin’s provided by incomplete preservation, a paucity of phylo- dilemma” regarding the origin and early evolution of Metazoa genetically informative characters, and uncertain expecta- arguably persists, because incontrovertible fossil evidence for tions of the anatomy of early animals, a number of animals remains largely, or some might say completely, absent Neoproterozoic fossils can reasonably be interpreted as from Neoproterozoic rocks [3]. -

Mrna Structure Dynamics Identifies the Embryonic RNA Regulome
bioRxiv preprint doi: https://doi.org/10.1101/274290; this version posted March 1, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. mRNA structure dynamics identifies the embryonic RNA regulome Jean-Denis Beaudoin1,*,‡, Eva Maria Novoa2,3,4,5,*, Charles E Vejnar1, Valeria Yartseva1, Carter Takacs1, Manolis Kellis2,3 and Antonio J Giraldez1,6,‡ RNA folding plays a crucial role in RNA function. However, our knowledge of the global structure of the transcriptome is limited to steady-state conditions, hindering our understanding of how RNA structure dynamics influences gene function. Here, we have characterized mRNA structure dynamics during zebrafish development. We observe that on a global level, translation guides structure rather than structure guiding translation. We detect a decrease in structure in translated regions, and we identify the ribosome as a major remodeler of RNA structure in vivo. In contrast, we find that 3’-UTRs form highly folded structures in vivo, which can affect gene expression by modulating miRNA activity. Furthermore, we find that dynamic 3’-UTR structures encode RNA decay elements, including regulatory elements in nanog and cyclin A1, key maternal factors orchestrating the maternal-to-zygotic transition. These results reveal a central role of RNA structure dynamics in gene regulatory programs. Introduction Previous studies have focused on the global analysis of RNA structures in cells at RNA carries out a broad range of functions,1 and steady-state. However, in an organism, critical RNA structure has emerged as a fundamental developmental and metabolic decisions are often regulatory mechanism, modulating various post- made when cells transition between cellular transcriptional events such as splicing2, states. -

Organizational Properties of a Functional Mammalian Cis-Regulome
bioRxiv preprint doi: https://doi.org/10.1101/550897; this version posted February 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Organizational properties of a functional mammalian cis-regulome Virendra K. Chaudhri, Krista Dienger-Stambaugh, Zhiguo Wu, Mahesh Shrestha and Harinder Singh*# Division of Immunobiology and Center for Systems Immunology, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio, 45220, USA #Current address: Center for Systems Immunology, Departments of Immunology and Computational and Systems Biology, The University of Pittsburgh, Pittsburgh, PA 15213 *Corresponding author Email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/550897; this version posted February 15, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Mammalian genomic states are distinguished by their chromatin and transcription profiles. Most genomic analyses rely on chromatin profiling to infer cis-regulomes controlling distinctive cellular states. By coupling FAIRE-seq with STARR-seq and integrating Hi-C we assemble a functional cis-regulome for activated murine B- cells. Within 55,130 accessible chromatin regions we delineate 9,989 active enhancers communicating with 7,530 promoters. The cis-regulome is dominated by long range enhancer-promoter interactions (>100kb) and complex combinatorics, implying rapid evolvability. Genes with multiple enhancers display higher rates of transcription and multi-genic enhancers manifest graded levels of H3K4me1 and H3K27ac in poised and activated states, respectively. Motif analysis of pathway-specific enhancers reveals diverse transcription factor (TF) codes controlling discrete processes.