Studies on the Genetics of Canine Hip Dysplasia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

This Thesis Has Been Submitted in Fulfilment of the Requirements for a Postgraduate Degree (E.G
This thesis has been submitted in fulfilment of the requirements for a postgraduate degree (e.g. PhD, MPhil, DClinPsychol) at the University of Edinburgh. Please note the following terms and conditions of use: This work is protected by copyright and other intellectual property rights, which are retained by the thesis author, unless otherwise stated. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the author. The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the author. When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given. ‘For the Good of the Breed’ Care, Ethics, and Responsibility in Pedigree Dog Breeding Chrissie Wanner PhD in Social Anthropology University of Edinburgh 2017 1 Declaration I declare that this thesis has been composed solely by myself and that it has not been submitted, in whole or in part, in any previous application for a degree. Except where states otherwise by reference or acknowledgment, the work presented is entirely my own. Signed: Date: 2 3 Abstract This thesis examines how the ethics of caring for pedigree dogs differ in the contexts of dog showing and veterinary practice. By highlighting conflicts around the shared use of ‘ordinary language’, I show how tensions between show‐world and veterinary perspectives relate to divergent understandings of ‘health’. Canine bodies speak to vets and breeders in conceptually different ways, so much so that breed‐specific features can be considered ‘perfect’ in the show‐ring yet ‘pathological’ in the veterinary clinic. -

Inbreeding Purge of Canine Hip and Elbow Dysplasia
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 2 August 2020 doi:10.20944/preprints202008.0027.v1 Article Effects of Long Term Selection in the Border Collie Dog Breed: Inbreeding Purge of Canine Hip and Elbow Dysplasia Virág Ács1, György Kövér2, János Farkas2, Árpád Bokor3, István Nagy4 1Department of Animal Nutrition, Kaposvár University, Kaposvár, H-7400, 40, Guba S. str., Hungary; 2Department of Mathematics and Informatics, Kaposvár University, Kaposvár, H-7400, 40, Guba S. str., Hungary; 3Department of Hippology, Kaposvár University, Kaposvár, H-7400, 40, Guba S. str., Hungary; 4Department of Animal Science, Kaposvár University, Kaposvár, H-7400, 40, Guba S. str., Hungary; *Corresponding author: [email protected] Simple Summary: For dog breeders, health is one of the main criteria when choosing a breeding animal, thus selection for good anatomy is the key to reduce orthopedic disorders. In many dog breeds, radiographic screening for canine hip and elbow dysplasia is a compulsory test for breeding, however, these multifactorial traits are determined by genetic and environmental factors. Therefore, it is really hard to eliminate these disorders from the population. In natural selection, such traits can “purge” out of the with inbreeding. The study aimed to examine the inbreeding-purge of canine hip and elbow dysplasia in the border collie breed. The main conclusion was, that with over-representation of homozygous individuals may have a positive effect on hip and elbow conformation. Abstract: Pedigree data of 13 339 border collie dog was collected along with hip and elbow dysplasia records (1352 CHD and 524 CED), and an inbreeding-purging (IP) model was created to detect possible purging. -

The Price of a Pedigree
The Price of a Pedigree DOG BREED STANDARDS AND BREED-RELATED ILLNESS The Price of a Pedigree: Dog breed standards and breed-related illness A report by Advocates for Animals 2006 Contents 1. Introduction: the welfare implications of pedigree dog breed standards 2. Current and future breeding trends 3. The prevalence of breed-related disease and abnormality 4. Breeds affected by hereditary hip and elbow dysplasia 4.1 The British Veterinary Association/Kennel Club hip and elbow dysplasia schemes 4.2 International studies of the prevalence of hip and elbow dysplasia 5. Breeds affected by inherited eye diseases 5.1 The British Veterinary Association/Kennel Club/ISDS Eye scheme 5.2 Further breed-related eye problems 6. Breeds affected by heart and respiratory disease 6.1 Brachycephalic Upper Airway Syndrome 6.2 Increased risk of heart conditions 7. Breed-related skin diseases 8. Inherited skeletal problems of small and long-backed breeds 8.1 Luxating patella 8.2 Intervertebral disc disease in chondrodystrophoid breeds 9. Bone tumours in large and giant dog breeds 10. Hereditary deafness 11. The Council of Europe and breed standards 11.1 Views of companion animal organisations on dog breeding 12. Conclusions and recommendations Appendix. Scientific assessments of the prevalence of breed-related disorders in pedigree dogs. Tables 1 – 9 and Glossaries of diseases References 1. Introduction: The welfare implications of pedigree dog breed standards ‘BREEDERS AND SCIENTISTS HAVE LONG BEEN AWARE THAT ALL IS NOT WELL IN THE WORLD OF COMPANION ANIMAL BREEDING.’ Animal Welfare, vol 8, 1999 1 There were an estimated 6.5 million dogs in the UK in 2003 and one in five of all households includes a dog.2 Only a minority (around a quarter) of these dogs are mongrels or mixed breed dogs. -
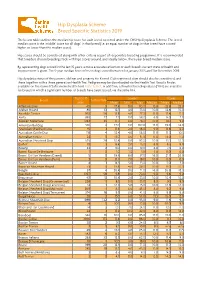
Hip Dysplasia Scheme Breed Specific Statistics 2019
Hip Dysplasia Scheme Breed Specific Statistics 2019 The below table outlines the median hip score for each breed screened under the CHS Hip Dysplasia Scheme. The breed median score is the ‘middle’ score for all dogs’ in that breed (i.e. an equal number of dogs in that breed have scored higher or lower than the median score). Hip scores should be considered along with other criteria as part of responsible breeding programme. It is recommended that breeders choose breeding stock with hips scores around, and ideally below, the 5-year breed median score. By representing dogs scored in the last 15 years, a more accurate reflection of each breed’s current state of health and improvement is given. The 5-year median here refers to dogs scored between 1st January 2015 and 31st December 2019. Hip dysplasia status of the parents, siblings and progeny for Kennel Club registered dogs should also be considered, and these together with a three generation Health Test Pedigree may be downloaded via the Health Test Results Finder, available on the Kennel Club's online health tool Mate Select. In addition, estimated breeding values (EBVs) are available for breeds in which a significant number of breeds have been scored, via the same link. Tested 15 15 years 5 years Breed Tested 2019 years Mean Min Max Median Mean Median Affenpinscher 40 0 17.9 8.0 90.0 13.0 23.8 23.0 Afghan Hound 85 33 12.3 4.0 73.0 10.0 12.6 10.0 Airedale Terrier 910 58 13.9 4.0 77.0 11.0 13.8 11.0 Akita 883 27 7.7 0.0 58.0 6.0 8.0 7.0 Alaskan Malamute 1242 25 11.7 0.0 78.0 10.0 10.1 9.0 -

AIS-Pennhip-Manual.Pdf
Training Manual Table of Contents Chapter 1: Introduction and Overview ............................................................................................... 5 Brief History of PennHIP ........................................................................................................................................5 Current Status of CHD ...........................................................................................................................................5 Requirements for Improved Hip Screening ............................................................................................................6 PennHIP Strategies ................................................................................................................................................7 The AIS PennHIP Procedure .................................................................................................................................8 AIS PennHIP Certification ......................................................................................................................................8 Purchasing a Distractor ..........................................................................................................................................9 Antech Imaging Services........................................................................................................................................9 Summary ............................................................................................................................................................ -

Registration Regulations Empowerment
New Zealand Kennel Club (Inc) (Affiliated with The Kennel Club, England) (Associated with the Federation Cynologique Internationale) REGISTRATION REGULATIONS (Reprinted with Additions and Amendments, to 1 April 2021) Headquarters Prosser Street, Porirua. New Zealand Kennel Club Private Bag 50903 Porirua 5240 Copyright - New Zealand Kennel Club (Inc.) Page | 1 CONTENTS Principles of the Registration System Section I - Registration Regulations Empowerment ........................................................................................................................ 4 Definitions .............................................................................................................................. 4 Charges, Fees, Forms and Signatures. ................................................................................. 4 The Executive Council, Registry and the Administration ........................................................ 5 The Register .......................................................................................................................... 5 Executive Council Powers ...................................................................................................... 6 Registry Details ...................................................................................................................... 6 Litter Notification .................................................................................................................... 6 Registration of Dogs. ............................................................................................................ -

A Retrospective Study on Findings of Canine Hip Dysplasia Screening in Kenya
Veterinary World, EISSN: 2231-0916 RESEARCH ARTICLE Available at www.veterinaryworld.org/Vol.8/November-2015/10.pdf Open Access A retrospective study on findings of canine hip dysplasia screening in Kenya Peter Kimeli1, Susan W. Mbugua1, Roger M. Cap2, Gilbert Kirui1, Tequiero O. Abuom1, Willy E. Mwangi1, Ambrose N. Kipyegon1 and John D. Mande1 1. Department of Clinical Studies, Faculty of Veterinary Medicine, University of Nairobi, P.O. Box 29053-00625, Kangemi, Kenya; 2. Sercombe Veterinary Surgeons, P.O Box 24878-00502, Nairobi, Kenya. Corresponding author: Peter Kimeli, e-mail: [email protected], SWM: [email protected], RMC: [email protected], GK: [email protected], TOA: [email protected], WEM: [email protected], ANK: [email protected], JDM: [email protected] Received: 15-06-2015, Revised: 27-09-2015, Accepted: 14-10-2015, Published online: 22-11-2015 doi: 10.14202/vetworld.2015.1326-1330 How to cite this article: Kimeli P, Mbugua SW, Cap RM, Kirui G, Abuom TO, Mwangi WE, Kipyegon AN, Mande JD (2015) A retrospective study on findings of canine hip dysplasia screening in Kenya, Veterinary World 8(11): 1326-1330. Abstract Aim: The current study was undertaken to evaluate the findings of canine hip dysplasia screening in Kenya. Materials and Methods: Records for 591 dogs were included in this study. The data was obtained from the national screening office, Kenya Veterinary Board, for the period between the years 1998 and 2014. Monthly screening records were assessed and information relating to year of evaluation, breed, sex, age, and hip score captured. Descriptive statistics of hip scores was computed based on year, sex, age, and breed. -

Heritability and Epidemiology of Canine Hip-Dysplasia Score and Its Components in Labrador Retrievers in the United Kingdom J.L.N
Preventive Veterinary Medicine 55 (2002) 95±108 Heritability and epidemiology of canine hip-dysplasia score and its components in Labrador retrievers in the United Kingdom J.L.N. Wood*, K.H. Lakhani, K. Rogers Animal Health Trust, Lanwades Park, Newmarket, Kentford CB8 7UU, Suffolk, UK Received 26 June 2001; accepted 26 June 2002 Abstract Hip-dysplasia (malformation of the coxofemoral joint) in dogs is a major health problem. Under the British Veterinary Association/Kennel Club's voluntary hip-dysplasia scheme, dog-owners/ breeders submit radiographs from animals >1-year-old, to ensure adequate skeletal maturity. An overall hip score quanti®es the degree of malformation in the hip joints of these animals, by summing the scores for nine components of the radiographs of both the left and right joints. The hip score data for 29,610 Labrador retrievers (registered with The Kennel Club, UK) were merged with the Kennel Club pedigree database for 472,435 Labrador retrievers. The merged data included the animal's identity, date of birth, sex and hip score and similar records for the dog's relatives, including the hip score if the relative had been tested. In recent years, breeding had been increasingly from tested parents. The mean hip score for male Labradors was signi®cantly higher than that for females. Regression modelling showed a signi®cant, positive dependence of the hip score of the offspring upon the hip scores of its sire, dam and grandparents. Genetic heritability (using data from 13,382 Labrador retrievers comprising 718 litters) was highly signi®cant: 0.34 from the two parents, 0.41 from sire alone and 0.30 from dam alone. -

Promoting Health of Dogs Through Breeding: the Finnish Kennel Club’S Tools
Promoting health of dogs through breeding: The Finnish Kennel Club’s tools Katariina Mäki, PhD Breeding expert 8.8.2014 [email protected] What does the Finnish Kennel Club do? • General breeding strategy • Breed-specific breeding strategies • Programme to combat hereditary diseases and defects (PEVISA) • Breeding database • Instructions to avoid exaggerated features • Dog registry guideline • Education for breeders and breeding councellors • Co-operation • National and international • Canine Health Research Fund Photo: T. Eerola Suomen Kennelliitto | Katariina Mäki Finnish Kennel Club’s rules and regulations • Developed with the health and wellbeing of dogs as primary concern • In harmony with the Animal Welfare Act and the Animal Welfare Decree as well as other official regulations that apply to the breeding of animals • Finnish Kennel Club’s (FKC) rules and regulations oblige all members, as well as people attending organized dog activities • By becoming a member one accepts also the rules Suomen Kennelliitto | Katariina Mäki Dog breeding in the Finnish Kennel Club • Breed associations and their subordinate clubs are responsible for directing the breeding in their respective breeds – Breeding goals and strategies • The FKC for example • Registers the dogs, making sure that the parents meet all the conditions • Maintains the Breeding database • Gives general breeding guidance and education Suomen Kennelliitto | Katariina Mäki Are we proceeding? How can you tell? If important traits of a breed are improving generation by generation as a result of selection and breeding, genetic improvement is taking place Photo: T. Eerola Suomen Kennelliitto | Katariina Mäki Hip dysplasia Bernese Mountain Dogs Bernese Mountain Dogs born 1980-1989 born 2000-2009 (mean 1,8 = almost C) (mean 1,3 = n. -

Hip Dysplasia in Dogs
Hip dysplasia in dogs The British Veterinary Association and the Kennel Club — working together for excellence in canine health Radiograph of normal hips in a greyhound. The hip is a ‘ball-and-socket’ joint, the ball being the head of the thigh bone or femur, and the socket being part of the pelvis called the acetabulum. In good hips the femoral head is smoothly rounded and fits tightly and deeply into the acetabulum. The outlines of the bone are clear since there is no secondary osteoarthrosis. Compare this image with those of dysplastic joints below and on page 3. ip dysplasia (HD) is a common from the muscles to drive the body forwards deformed joint results in varying amounts of inherited orthopaedic problem with maximum strength and speed. It is the inflammation and degeneration which lead to of dogs and a wide number close relationship of the femoral head (ball) more deformity. This progressive deformation of other mammals. Abnormal and acetabulum (socket) which enables rapid is sometimes referred to as remodelling. Hdevelopment of the structures that make changes of direction. The entire hip joint is a Some dogs may treble their size and body up the hip joint leads to subsequent joint unit comprising the bony structures contained weight in just three months of adolescence deformity. ‘Dysplasia’ means abnormal within a joint capsule and supported by so it is not surprising that there are many growth. The developmental changes ligaments, tendons and muscles together critical factors for the puppy at this stage. appear first and because they are related with all their blood vessels and nerves. -

Hip Dysplasia: AVA/ANKC Hip Score Scheme
Hip Dysplasia: AVA/ANKC Hip Score Scheme Hip dysplasia (HD) is a common inherited orthopaedic problem of dogs and a wide number of other mammals. Developmental demands Abnormal development of the structures that make up It is argued that dogs are not born with hip joints already the hip joint leads to subsequent joint deformity. affected by dysplasia (unlike humans) but that any faults ‘Dysplasia’ means abnormal growth. The developmental in development will tend to escalate with time, changes appear first and because they are related to particularly during the rapid growth phase from about 14 growth, they are termed primary changes. Subsequently to 26 weeks of age. However, changes begin as the very these changes may lead to excessive wear and tear. The young puppy starts to become active and continue until secondary changes may be referred to as osteoarthritis the puppy is skeletally mature. Wear and tear of the (OA), osteoarthrosis or degenerative joint disease (DJD). deformed joint results in varying amounts of Later one or both hip joints may become mechanically inflammation and degeneration which lead to more defective. At this stage the joint(s) may be painful and deformity. This progressive deformation is sometimes cause lameness. In extreme cases the dog may find referred to as remodelling. movement very difficult and may suffer considerably. Some dogs may treble their size and body weight in just three months of adolescence so it is not surprising that there are many critical factors for the puppy at this stage. All the essential nutritional requirements for skeletal Structure and function growth must be available in the right proportions and at the right time. -

Hip Dysplasia and What Price a Normal Hip
Veterinary Information Sheet Hip Dysplasia and What Price a Normal Hip Dr Karen Hedberg BVSc. - 2012. Introduction The following article is an attempt to cover the many and varied aspects of hip dysplasia; its definition, the factors affecting the severity of the signs seen, the treatment of HD as well as the control of HD by (a) various schemes and (b) the genetic aspects. This somewhat rambling account is an attempt to show that the control of HD by concerned breeders is both difficult and complex. Additionally, the general public is being increasingly told that a “normal” hip is the only one acceptable, and anything above that may require surgical intervention. As both a breeder and a veterinarian, we need to look at this problem from all angles and present to the general public a more realistic view of the condition, not only for ourselves, but for all breeds where clubs are trying to lower the severity of the HD problem. As the general public is becoming far more litigious in these matters it behoves us to present a rational approach with realistic goals. Breeders' Aims When we are breeding dogs, in addition to producing better show animals, we should also be trying to breed as sound an animal as possible. This encompasses all of the following:- physical, mental and genetic soundness as well as breed type (ie. it must still resemble the breed!). All of these areas are of great importance, some are weighted more heavily than others in different breeds. Over time various areas come under heavier pressure, eg.