Glutamate Addiction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ie-O-JY -CH2 S (56) References Cited Wherein N Is 0 to 6, X" and Y Are Independently H, Cl, F, CH, CH, CH, CH, OCH, OH, CF, OCF, NO, U.S
US005574060A United States Patent (19) 11) Patent Number: 5,574,060 Rothman et al. (45) Date of Patent: Nov. 12, 1996 54 SELECTIVE INHIBITORS OF BIOGENIC "Hydrindene Derivatives as Potential Oral Hypoglycemic AMINE TRANSPORTERS Agents: N-Alkyl 1,2,3,3a,4,8b-Hexahydroindeno1,2-b) pyrroles', De et al., Journal of Pharmaceutical Sciences, vol. (75) Inventors: Richard Rothman, Silver Spring, Md.; 62, No. 8, Aug. 1973, pp. 1363-1364. Frank J. Carroll, Durham, N.C.; Bruce Blough, Raleigh, N.C.; Samuel (List continued on next page.) W. Mascarella, Hillsborough, N.C. Primary Examiner-Mark L. Berch (73) Assignees: The United States of America as Attorney, Agent, or Firm-Morgan & Finnegan LLP represented by the Department of 57) ABSTRACT Health and Human Services, Washington, D.C.; Office of Technology The present invention provides a compound having the Transfer, Bethesda, Md.; Research Structure: Triangle Institue, Research Triangle Park, N.C. (21) Appl. No.: 203,222 22 Filed: Feb. 28, 1994 Related U.S. Application Data H 63 Continuation-in-part of Ser. No. 105,747, Aug. 12, 1993, abandoned. wherein X, Y, and Z are independently H, Cl, Br, F, OCH, I, or an alkyl group having 1 to 6 carbon atoms; and R is (51) Int. Cl." .......................... A61K 31/40; A61K 31/47; C07D 403/06; C07D 471/08 X (52) U.S. Cl. ............................ 514/411; 546/94; 546/146; 54.6/150; 548/427: 548/428 58) Field of Search .............................. 548/427; 514/411 -ie-O-JY -CH2 S (56) References Cited wherein n is 0 to 6, X" and Y are independently H, Cl, F, CH, CH, CH, CH, OCH, OH, CF, OCF, NO, U.S. -

Can N-Methyl-D-Aspartate Receptor Hypofunction in Schizophrenia Be Localized to an Individual Cell Type?
REVIEW published: 21 November 2019 doi: 10.3389/fpsyt.2019.00835 Can N-Methyl-D-Aspartate Receptor Hypofunction in Schizophrenia Be Localized to an Individual Cell Type? Alexei M. Bygrave 1, Kasyoka Kilonzo 2, Dimitri M. Kullmann 3, David M. Bannerman 4 and Dennis Kätzel 2* 1 Department of Neuroscience, Johns Hopkins University, Baltimore, MD, United States, 2 Institute of Applied Physiology, Ulm University, Ulm, Germany, 3 UCL Queen Square Institute of Neurology, University College London, London, United Kingdom, 4 Department of Experimental Psychology, University of Oxford, Oxford, United Kingdom Hypofunction of N-methyl-D-aspartate glutamate receptors (NMDARs), whether caused by endogenous factors like auto-antibodies or mutations, or by pharmacological or genetic manipulations, produces a wide variety of deficits which overlap with—but do not precisely match—the symptom spectrum of schizophrenia. In order to understand how NMDAR hypofunction leads to different components of the syndrome, it is necessary to take into account which neuronal subtypes are particularly affected by it in terms of detrimental Edited by: functional alterations. We provide a comprehensive overview detailing findings in rodent Bernat Kocsis, Harvard Medical School, models with cell type–specific knockout of NMDARs. Regarding inhibitory cortical cells, United States an emerging model suggests that NMDAR hypofunction in parvalbumin (PV) positive Reviewed by: interneurons is a potential risk factor for this disease. PV interneurons display a selective Margarita Behrens, Salk Institute for Biological Studies, vulnerability resulting from a combination of genetic, cellular, and environmental factors United States that produce pathological multi-level positive feedback loops. Central to this are two Vasileios Kafetzopoulos, antioxidant mechanisms—NMDAR activity and perineuronal nets—which are themselves Harvard Medical School, United States impaired by oxidative stress, amplifying disinhibition. -

Cerebellar Toxicity of Phencyclidine
The Journal of Neuroscience, March 1995, 75(3): 2097-2108 Cerebellar Toxicity of Phencyclidine Riitta N&kki, Jari Koistinaho, Frank Ft. Sharp, and Stephen M. Sagar Department of Neurology, University of California, and Veterans Affairs Medical Center, San Francisco, California 94121 Phencyclidine (PCP), clizocilpine maleate (MK801), and oth- Phencyclidine (PCP), dizocilpine maleate (MK801), and other er NMDA antagonists are toxic to neurons in the posterior NMDA receptor antagonistshave attracted increasing attention cingulate and retrosplenial cortex. To determine if addition- becauseof their therapeutic potential. These drugs have neuro- al neurons are damaged, the distribution of microglial ac- protective properties in animal studies of focal brain ischemia, tivation and 70 kDa heat shock protein (HSP70) induction where excitotoxicity is proposedto be an important mechanism was studied following the administration of PCP and of neuronal cell death (Dalkara et al., 1990; Martinez-Arizala et MK801 to rats. PCP (10-50 mg/kg) induced microglial ac- al., 1990). Moreover, NMDA antagonists decrease neuronal tivation and neuronal HSP70 mRNA and protein expression damage and dysfunction in other pathological conditions, in- in the posterior cingulate and retrosplenial cortex. In ad- cluding hypoglycemia (Nellgard and Wieloch, 1992) and pro- dition, coronal sections of the cerebellar vermis of PCP (50 longed seizures(Church and Lodge, 1990; Faingold et al., 1993). mg/kg) treated rats contained vertical stripes of activated However, NMDA antagonists are toxic to certain neuronal microglial in the molecular layer. In the sagittal plane, the populations in the brain. Olney et al. (1989) demonstratedthat microglial activation occurred in irregularly shaped patch- the noncompetitive NMDA antagonists,PCP, MK801, and ke- es, suggesting damage to Purkinje cells. -

RTI-4793-14, a New Ligand with High Affinity and Selectivity For
SYNAPSE 16:59-65 (1994) RTI-4793-14, a New Ligand With High Affinity and Selectivity for the ( +)-MK801-Insensitive [3H]1 -[ 1 -( 2- thienyl)cyclohexyl]piperidine Binding Site (PCP Site 2) of Guinea Pig Brain CARL B. GOODMAN, D. NIGEL THOMAS, AGU PERT, BETSEY EMILIEN, JEAN L. CADET, F. IVY CARROLL, BRUCE E. BLOUGH, S. WAYNE MASCARELLA, MICHAEL A. ROGAWSKI, SWAMINATHAN SUBRAMANIAM, AM) RICHARD B. ROTHMAN Clinical Psychopharmacology Section, NIDMNIH Addiction Research Center, Baltimore, Maryland 21224 (C.B.G., B.E., J.L.C., R.B.R.); Biological Psychiatry Branch, NZMH (D.N.T., A.P.) and Neuronal Excitability Section, Epilepsy Research Branch, NINDS (M.A.R., S.S.), NIH, Bethesda, Maryland 20892; and Chemistry and Life Sciences, Organic and Medicinal Chemistry, Research Triangle Institute, Research Triangle Park, North Carolina 27709-2194 (F.I.C., B.E.B., S.W.M.) KEY WORDS Phencyclidine receptor, RTI-4793-14, PCP, Biogenic amine reuptake carrier, ( + )-MK801, Guinea pig brain ABSTRACT L3HlTCP, an analog of the dissociative anesthetic phencyclidine (PCP), binds with high affinity to two sites in guinea pig brain membranes, one that is MK-801 sensitive and one that is not. The MK-801-sensitive site (PCP site 1) is associated with NMDA receptors, whereas the MK-801-insensitive site (PCP site 2) may be associated with biogenic amine transporters (BAT).Although several “BAT ligands” are known that bind selectively to PCP site 2 and not to PCP site 1 (such as indatraline), these corn- pounds have low affinity for site 2 (K, values > 1 pM). Here we demonstrate that the novel pyrrole RTI-4793-14 is a selective, high affinity ligand for PCP site 2. -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Stems for Nonproprietary Drug Names
USAN STEM LIST STEM DEFINITION EXAMPLES -abine (see -arabine, -citabine) -ac anti-inflammatory agents (acetic acid derivatives) bromfenac dexpemedolac -acetam (see -racetam) -adol or analgesics (mixed opiate receptor agonists/ tazadolene -adol- antagonists) spiradolene levonantradol -adox antibacterials (quinoline dioxide derivatives) carbadox -afenone antiarrhythmics (propafenone derivatives) alprafenone diprafenonex -afil PDE5 inhibitors tadalafil -aj- antiarrhythmics (ajmaline derivatives) lorajmine -aldrate antacid aluminum salts magaldrate -algron alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol combined alpha and beta blockers labetalol medroxalol -amidis antimyloidotics tafamidis -amivir (see -vir) -ampa ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) subgroup: -ampanel antagonists becampanel -ampator modulators forampator -anib angiogenesis inhibitors pegaptanib cediranib 1 subgroup: -siranib siRNA bevasiranib -andr- androgens nandrolone -anserin serotonin 5-HT2 receptor antagonists altanserin tropanserin adatanserin -antel anthelmintics (undefined group) carbantel subgroup: -quantel 2-deoxoparaherquamide A derivatives derquantel -antrone antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine antineoplastics (arabinofuranosyl derivatives) fazarabine fludarabine aril-, -aril, -aril- antiviral (arildone derivatives) pleconaril arildone fosarilate -arit antirheumatics (lobenzarit type) lobenzarit clobuzarit -arol anticoagulants (dicumarol type) dicumarol -

Modifications to the Harmonized Tariff Schedule of the United States To
U.S. International Trade Commission COMMISSIONERS Shara L. Aranoff, Chairman Daniel R. Pearson, Vice Chairman Deanna Tanner Okun Charlotte R. Lane Irving A. Williamson Dean A. Pinkert Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Modifications to the Harmonized Tariff Schedule of the United States to Implement the Dominican Republic- Central America-United States Free Trade Agreement With Respect to Costa Rica Publication 4038 December 2008 (This page is intentionally blank) Pursuant to the letter of request from the United States Trade Representative of December 18, 2008, set forth in the Appendix hereto, and pursuant to section 1207(a) of the Omnibus Trade and Competitiveness Act, the Commission is publishing the following modifications to the Harmonized Tariff Schedule of the United States (HTS) to implement the Dominican Republic- Central America-United States Free Trade Agreement, as approved in the Dominican Republic-Central America- United States Free Trade Agreement Implementation Act, with respect to Costa Rica. (This page is intentionally blank) Annex I Effective with respect to goods that are entered, or withdrawn from warehouse for consumption, on or after January 1, 2009, the Harmonized Tariff Schedule of the United States (HTS) is modified as provided herein, with bracketed matter included to assist in the understanding of proclaimed modifications. The following supersedes matter now in the HTS. (1). General note 4 is modified as follows: (a). by deleting from subdivision (a) the following country from the enumeration of independent beneficiary developing countries: Costa Rica (b). -

Pdf4 Complex I and the Aryl Palladium Precursor II Underwent Sequential Single Electron Abstraction from Aryl Pd(II) Complex
Design, synthesis, methodology development, and evaluation of PET imaging agents targeting cancer and CNS disorders By Gengyang Yuan B.S. in Chemical Engineering and Technology, Zhejiang University of Technology M.S. in Pharmaceutical Engineering, Zhejiang University A dissertation submitted to The Faculty of the College of Science of Northeastern University in partial fulfillment of the requirements for the degree of Doctor of Philosophy April 21, 2017 Dissertation directed by Michael P. Pollastri Associate Professor and Chair of Chemistry and Chemical Biology Co-directed by Neil Vasdev Adjunct Associate Professor of Chemsitry and Chemical Biology Associate Professor of Radiology, Massachusetts General Hospital and Harvard Medical School Dedication To my parents Zhijun and Yongmian and my wife Ran and daughter Isabella ii Acknowledgements This dissertation would not have been possible without the support, guidance and encouragement of numerous people who have helped me along the way. First and foremost, I would like to thank Northeastern University and the Department of Chemistry and Chemical Biology for supporting me to pursue my doctoral study. I would like to especially thank my current advisor Professor Michael Pollastri for helping me out when I needed it the most. I appreciate you for taking me into your group and giving me full support to finish my thesis projects. I also especially thank my co-advisor Professor Neil Vasdev for taking me into his group at Mass. General Hospital & Harvard Medical School and teaching me the PET radiochemistry and PET imaging. I could not image how I could accomplish this work without your help. I also got a lot of help from Dr. -

United States Patent (10 ) Patent No.: US 10,660,887 B2 Javitt (45 ) Date of Patent: *May 26 , 2020
US010660887B2 United States Patent (10 ) Patent No.: US 10,660,887 B2 Javitt (45 ) Date of Patent : *May 26 , 2020 (54 ) COMPOSITION AND METHOD FOR (56 ) References Cited TREATMENT OF DEPRESSION AND PSYCHOSIS IN HUMANS U.S. PATENT DOCUMENTS 6,228,875 B1 5/2001 Tsai et al. ( 71 ) Applicant : Glytech , LLC , Ft. Lee , NJ (US ) 2004/0157926 A1 * 8/2004 Heresco - Levy A61K 31/198 514/561 (72 ) Inventor: Daniel C. Javitt , Ft. Lee , NJ (US ) 2005/0261340 Al 11/2005 Weiner 2006/0204486 Al 9/2006 Pyke et al . 2008/0194631 Al 8/2008 Trovero et al. ( 73 ) Assignee : GLYTECH , LLC , Ft. Lee , NJ (US ) 2008/0194698 A1 8/2008 Hermanussen et al . 2010/0069399 A1 * 3/2010 Gant CO7D 401/12 ( * ) Notice : Subject to any disclaimer, the term of this 514 / 253.07 patent is extended or adjusted under 35 2010/0216805 Al 8/2010 Barlow 2011/0207776 Al 8/2011 Buntinx U.S.C. 154 ( b ) by 95 days . 2011/0237602 A1 9/2011 Meltzer This patent is subject to a terminal dis 2011/0306586 Al 12/2011 Khan claimer . 2012/0041026 A1 2/2012 Waizumi ( 21 ) Appl. No.: 15 /650,912 FOREIGN PATENT DOCUMENTS CN 101090721 12/2007 ( 22 ) Filed : Jul. 16 , 17 KR 2007 0017136 2/2007 WO 2005/065308 7/2005 (65 ) Prior Publication Data WO 2005/079756 9/2005 WO 2011044089 4/2011 US 2017/0312275 A1 Nov. 2 , 2017 WO 2012/104852 8/2012 WO 2005/000216 9/2013 WO 2013138322 9/2013 Related U.S. Application Data (63 ) Continuation of application No. -

High-Throughput Brain Activity Mapping and Machine Learning As a Foundation for Systems Neuropharmacology
High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology Lin, Xudong; Duan, Xin; Jacobs, Claire; Ullmann, Jeremy; Chan, Chung-Yuen; Chen, Siya; Cheng, Shuk-Han; Zhao, Wen-Ning; Poduri, Annapurna; Wang, Xin; Haggarty, Stephen J.; Shi, Peng Published in: Nature Communications Published: 03/12/2018 Document Version: Final Published version, also known as Publisher’s PDF, Publisher’s Final version or Version of Record License: CC BY Publication record in CityU Scholars: Go to record Published version (DOI): 10.1038/s41467-018-07289-5 Publication details: Lin, X., Duan, X., Jacobs, C., Ullmann, J., Chan, C-Y., Chen, S., Cheng, S-H., Zhao, W-N., Poduri, A., Wang, X., Haggarty, S. J., & Shi, P. (2018). High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology. Nature Communications, 9, [5142]. https://doi.org/10.1038/s41467-018-07289- 5 Citing this paper Please note that where the full-text provided on CityU Scholars is the Post-print version (also known as Accepted Author Manuscript, Peer-reviewed or Author Final version), it may differ from the Final Published version. When citing, ensure that you check and use the publisher's definitive version for pagination and other details. General rights Copyright for the publications made accessible via the CityU Scholars portal is retained by the author(s) and/or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Users may not further distribute the material or use it for any profit-making activity or commercial gain. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -
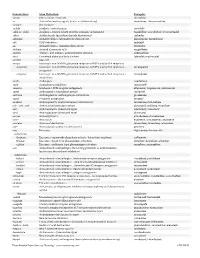
Downloaded from Survive Nursing | Survivenursing.Com V20110426
Generic Stem Stem Definition Examples -abine (see -arabine, -citabine) decitabine -ac Anti-inflammatory agents (acetic acid derivatives) bromfenac; dexpemedolac -acetam See -racetam -actide Synthetic corticotropins seractide -adol or -aldol- Analgesics (mixed opiate receptor agonists/ antagonists) tazadolene; spiradolene; levonantradol -adox Antibacterials (quinoline dioxide derivatives) carbadox -afenone Antiarrhythmics (propafenone derivatives) alprafenone; diprafenone -afil PDE5 inhibitors tadalafil -aj- Antiarrhythmics (ajmaline derivatives) lorajmine -aldrate Antacid aluminum salts magaldrate -algron Alpha1 - and alpha2 - adrenoreceptor agonists dabuzalgron -alol Combined alpha and beta blockers labetalol; medroxalol -amivir (see -vir) -ampa Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) -ampanel Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; becampanel antagonists -ampator Ionotropic non-NMDA glutamate receptors (AMPA and/or KA receptors) ; forampator modulators -andr- Androgens nandrolone -anib Angiogenesis inhibitors semaxanib -anserin Serotonin 5-HT2 receptor antagonists altanserin; tropanserin; adatanserin -antel Anthelmintics (undefined group) carbantel -antrone Antineoplastics; anthraquinone derivatives pixantrone -apsel P-selectin antagonists torapsel -arabine Antineoplastics (arabinofuranosyl derivatives) fazarabine; fludarabine aril-, -aril, -aril- Antiviral (arildone derivatives) pleconaril; arildone; fosarilate -arit Antirheumatics (lobenzarit type) lobenzarit; clobuzarit -arol