Technical Considerations for Pen, Jet, and Related Injectors Intended for Use with Drugs and Biological Products
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Incremental Costs of Introducing Jet Injection Technology for Delivery of Routine Childhood Vaccinations: Comparative Analysis from Brazil, India, and South Africa
Vaccine 29 (2011) 969–975 Contents lists available at ScienceDirect Vaccine journal homepage: www.elsevier.com/locate/vaccine Incremental costs of introducing jet injection technology for delivery of routine childhood vaccinations: Comparative analysis from Brazil, India, and South Africa Ulla K. Griffiths a,∗, Andreia C. Santos a, Neeti Nundy b, Erica Jacoby b, Dipika Matthias b a London School of Hygiene and Tropical Medicine, Tavistock Place, London WC1H 9SH, UK b PATH, 2201 Westlake Avenue, Suite 200, Seattle, WA 98121, USA article info abstract Article history: Background: Disposable-syringe jet injectors (DSJIs) have the potential to deliver vaccines safely and Received 14 May 2010 affordably to millions of children around the world. We estimated the incremental costs of transitioning Received in revised form 9 November 2010 from needles and syringes to delivering childhood vaccines with DSJIs in Brazil, India, and South Africa. Accepted 15 November 2010 Methods: Two scenarios were assessed: (1) DSJI delivery of all vaccines at current dose and depth; (2) a Available online 27 November 2010 change to intradermal (ID) delivery with DSJIs for hepatitis B and yellow fever vaccines, while the other vaccines are delivered by DSJIs at current dose and depth. The main advantage of ID delivery is that only Keywords: a small fraction of the standard dose may be needed to obtain an immune response similar to that of Vaccine Jet injector subcutaneous or intramuscular injection. Cost categories included were vaccines, injection equipment, Costs waste management, and vaccine transport. Some delivery cost items, such as training and personnel Brazil were excluded as were treatment cost savings caused by a reduction in diseases transmitted due to India unsafe injections. -

Administering a Subcutaneous Injection
Inject medication slowly and release skin Will the injection hurt? Children may describe a pinching/stinging or bee-sting sensation during Our Lady’s Children’s Leave needle in place for 5-10 seconds after and just after the injection. It is normal for the Hospital, Crumlin, Dublin injecting medication, if possible injection site to be a little red and tender. It is 12 Remove needle swiftly and smoothly expected that children may be afraid of injections. It is important to be honest and explain the Wipe area gently with cotton wool, do not rub injection in a manner that they can understand. ….where children’s health comes first as this may be sore. Apply a plaster if needed. How to reduce any discomfort: ADVICE FOR • Prepare your child, explain why this is necessary PARENTS/GUARDIANS/CHILDREN and how you will give the injection Step 4. Dispose of the Needle/Syringe • Use distraction and play to amuse your child Administering a • Encourage them to practice on their teddy or doll. For infants, give them a soother/comforter if Subcutaneous Injection Dispose of needle and syringe immediately into they use one a sharps bin • Ensure clothing over the injection site is not tight My child has a bleeding disorder: seek medical advice before giving injections to your child. Auto-injector: Some children use an automatic injection device, often called a ‘rocket’. Specific Instructions: ……………………………………………………………………… ………………………………………………………………………. Developed by: Naomi Bartley, Clinical Placement Coordinator. CONTACT DETAILS Example of an Auto-Injector st Date issued: July 2014, October 2012 : 1 edn. Ward / Dept. Name: _________________ Date of review: July 2017 Telephone: ___________________ Follow the manufacture’s instructions for ©2014, Our Lady’s Children’s Hospital Crumlin, Dublin 12. -

For Nexplanon® Insertion and Removal G
Wilson et al. Contraception and Reproductive Medicine (2020) 5:1 Contraception and https://doi.org/10.1186/s40834-020-00104-x Reproductive Medicine RESEARCH Open Access Comparison of traditional anesthesia method and jet injector anesthesia method (MadaJet XL®) for Nexplanon® insertion and removal G. Anthony Wilson* , Julie W. Jeter, William S. Dabbs, Amy Barger Stevens, Robert E. Heidel and Shaunta’ M. Chamberlin Abstract Background: This study compared a needle-free anesthesia method with traditional local anesthesia for insertion and removal of Nexplanon® long-acting removable contraceptive device. In our clinic, patients often avoid this highly effective form of contraception due to fear of needles. We sought to determine if patients perceived a difference in pain with the injection, anxiety level or pain with the procedure when local anesthesia was given with a needle v/s a needle-free jet injector device. Methods: Patients were randomly assigned to one of two groups: jet injector or needle lidocaine delivery. Outcomes were ease of use, patient anxiety level, painfulness, and efficacy of anesthesia method. Results: Patient pain perception with administration of jet injector lidocaine was statistically lower than traditional needle with no difference in anxiety or ease of use, or efficacy of the anesthesia. Conclusion: The jet injector device is a reasonable alternative to needle injection delivery of anesthesia prior to insertion/removal of Nexplanon® device. Further studies may determine whether this needle-free alternative for administration of local anesthetic would result in more women choosing Nexplanon® as a contraceptive method. Keywords: Local anesthetic, Nexplanon®, Patient anxiety Background injection of lidocaine differed between the methods of As with many procedures [1, 2], patients often cite a fear delivery, and whether the presence or absence of needles of needles as a major reason to decline Nexplanon® in the anesthesia method affected patient anxiety level. -

Fascia Iliaca Block in the Emergency Department for Hip Fracture
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Serveur académique lausannois Pasquier et al. BMC Geriatrics (2019) 19:180 https://doi.org/10.1186/s12877-019-1193-0 RESEARCH ARTICLE Open Access Fascia iliaca block in the emergency department for hip fracture: a randomized, controlled, double-blind trial Mathieu Pasquier1, Patrick Taffé2, Olivier Hugli1, Olivier Borens3, Kyle Robert Kirkham4 and Eric Albrecht5* Abstract Background: Hip fracture causes moderate to severe pain and while fascia iliaca block has been reported to provide analgesic benefit, most previous trials were unblinded, with subsequent high risks of performance, selection and detection biases. In this randomized, control double-blind trial, we tested the hypothesis that a fascia iliaca block provides effective analgesia for patients suffering from hip fracture. Methods: Thirty ASA I-III hip fracture patients over 70 years old, who received prehospital morphine, were randomized to receive either a fascia iliaca block using 30 ml of bupivacaine 0.5% with epinephrine 1:200,000 or a sham injection with normal saline. The fascia iliaca block was administered by emergency medicine physicians trained to perform an anatomic landmark-based technique. The primary outcome was the comparison between groups of the longitudinal pain score profiles at rest over the first 45 min following the procedure (numeric rating scale, 0–10). Secondary outcomes included the longitudinal pain score profiles on movement and the comparison over 4 h, 8 h, 12 h, and 24 h after the procedure, along with cumulative intravenous morphine consumption at 24 h. Results: At baseline, the fascia iliaca group had a lower mean pain score than the sham injection group, both at rest (difference = − 0.9, 95%CI [− 2.4, 0.5]) and on movement (difference = − 0.9, 95%CI [− 2.7; 0.9]). -

Growth Hormone Treatment Without a Needle Using the Preci-Jet 50
Archives of Disease in Childhood 1997;76:65–67 65 Growth hormone treatment without a needle Arch Dis Child: first published as 10.1136/adc.76.1.65 on 1 January 1997. Downloaded from using the Preci-Jet 50 transjector P Bareille, M MacSwiney, A Albanese, C De Vile, R Stanhope Abstract Patients and methods A new delivery system (Preci-Jet 50) Twenty eight patients (11 girls, 17 boys) were which administers growth hormone randomly selected from the endocrine clinic at through the skin using high pressure and Great Ormond Street Hospital for Children. without a needle was evaluated. This All patients were receiving growth hormone device was inconvenient and painful com- treatment, 19 through an autoinjector and nine pared with a pen injection system. The with needles and syringes. None was using a conclusion is that the Preci-Jet is not the pen injector system at that time but nine had used one previously. The exclusion criteria panacea for solving the problem of com- were bleeding disorders and diseases of colla- pliance with subcutaneous growth hor- gen synthesis. Mean age was 10.1 (range mone injections. 5.7–16.7) years. The growth hormone dose ( 1997;76:65–67) Arch Dis Child range was from 15 to 30 IU/m2 body surface area/week. The subjects were randomly subdi- Keywords: growth hormone treatment; transcutaneous high pressure administration (jet); needle phobia. vided into two groups, A and B. Sex distribu- tion was similar in both groups but mean age diVered, with values of 8.7 and 11.2 years in groups A and B respectively. -
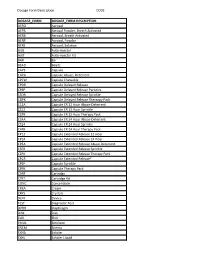
Dosage Form Description CODE
Dosage Form Description CODE DOSAGE_FORM DOSAGE_FORM DESCRIPTION AERO Aerosol AEPB Aerosol Powder, Breath Activated AERB Aerosol, Breath Activated AERP Aerosol, Powder AERS Aerosol, Solution AUIJ Auto-injector AJKT Auto-injector Kit BAR Bar BEAD Beads CAPS Capsule CAPA Capsule Abuse- Deterrent CPCW Capsule Chewable CPDR Capsule Delayed Release CPEP Capsule Delayed Release Particles CSDR Capsule Delayed Release Sprinkle CDPK Capsule Delayed Release Thereapy Pack C12A Capsule ER 12 Hour Abuse-Deterrent CS12 Capsule ER 12 Hour Sprinkle C2PK Capsule ER 12 Hour Therapy Pack C24A Capsule ER 24 Hour Abuse-Deterrent CS24 Capsule ER 24 Hour Sprinkle C4PK Capsule ER 24 Hour Therapy Pack CP12 Capsule Extended Release 12 Hour CP24 Capsule Extended Release 24 Hour CPEA Capsule Extended Release Abuse-Deterrent CSER Capsule Extended Release Sprinkle CEPK Capsule Extended Release Therapy Pack CPCR Capsule Extended Release* CPSP Capsule Sprinkle CPPK Capsule Therapy Pack CART Cartridge CTKT Cartridge Kit CONC Concentrate CREA Cream CRYS Crystals DEVI Device TEST Diagnostic Test DPRH Diaphragm DISK Disk ELIX Elixir EMUL Emulsion ENEM Enema EXHA Exhaler EXHL Exhaler Liquid Dosage Form Description CODE DOSAGE_FORM DOSAGE_FORM DESCRIPTION EXHP Exhaler Powder EXHS Exhaler Solution EXHU Exhaler Suspension FILM Film FLAK Flakes EXTR Fluid Extract FOAM Foam GAS Gas GEL Gel SOLG Gel Forming Solution GRAN Granules GREF Granules Effervescent GUM Gum IMPL Implant INHA Inhaler INJ Injectable INST Insert IUD Intrauterine Device JTAJ Jet-injector (Needleless) JTKT Jet-injector -

Cold and Flu Season
The DrugSmith is The Monthly Newsletter of The Smith Drug Company A Division of J M Smith Corporation Spartanburg, SC • Paragould, AR • Valdosta, GA Cold and Flu Season National Flu Vaccination Week World AIDS Month Introducing: Inject-Safe™ Barrier Bandage The first and only bandage designed specifically for injections. • Improved Safety Relieving Symptoms of the Common Cold 4 • Lower Cost National Influenza Vaccination Week 2016 6 • More Convenient • Less Pain What's New With The Flu? 7 Why Get A Flu Shot? 9 HealthWise Connect: Coming Soon! 11 World AIDS Day 12 Colon Rectal Cancer: Basic Facts 16 Healthwise™ Clinical Solutions Healthwise™ Circular Program DollarWise™ Program Good Sense® Controlled Label Program Greeting Cards Program Continuing Education Gift and Trade Show ™ Benefits of the Inject-Safe™ Barrier Bandage Rx QuickShip Third Party Station Inject-Safe™ Barrier Bandages self-seal to help contain Pharmacy First bleeding following an injection. DrugSmith™ Monthly Newsletter Consistent with “Universal Precautions” established by Smith Weekly e-Blast OSHA. Diabeticare Program Hamacher Retail Zone Pricing Protects the healthcare provider from coming into contact with potential blood borne pathogens. Home Health Care Catalog HealthWise Signage Program Allows healthcare provider to use both hands to dispose of Smith Gift Box Gift Category the needle. Vials and Vitamin Program Meets OSHA Blood borne Pathogen Standard's definition Direct Mail Advertising of "Engineering Control," for use to reduce employee Well Staffed Customer Service -

Refi Ning Procedures for the Administration of Substances
WORKINGPARTY REPORT Rening procedures for theadministration of substances Report of the BVAAWF =FRAME=RSPCA=UFAW JointWorking Group on Renement Members of theJoint Working Groupon Renement: D.B.Morton (Chairman), M.Jennings (Secretary),A. Buckwell, R.Ewbank, C.Godfrey, B.Holgate, I. Inglis, R.James, C.Page,I. Sharman, R.Verschoyle, L.Westall &A.B.Wilson Contents Preface 2 3.6 Intraperitoneal 19 1Introduction and aimsof thereport 2 3.7 Intratracheal 20 3.8 Intravaginal 20 2Generalprinc iplesof `good practice’ 3 2.1 Planningand preparation 3 3.9 Intravenous and intra-arterial 21 2.1.1 Experimental aims 3 3.10 Oral routes 25 2.1.2 Theroute 3 3.10.1 Inclusionin an animal’s food orwater 25 2.1.3 Thesubstance 3 3.10.2 Dosingdirectly into 2.1.4 Theanimal 5 the pharynx 27 2.1.5 Thetechnique 7 3.10.3 Oral gavage 28 2.1.6 Staff and training 7 3.11 Osmotic minipumps 30 2.2 Technical preparation 3.12 Respiratory routes 31 andaftercare 8 3.12.1 Wholebody exposure 31 2.3 Generalre® nem entfor 3.12.2 Noseonly =Snout only all routes 9 exposure 32 3Re®nem entfor individual routes 3.12.3 Mask exposure 33 and procedures 13 3.13 Subcutaneous 34 3.1 Intra-articular 13 3.14 TopicalÐderm al 35 3.2 Intracerebral (intracerebro- 3.15 TopicalÐoc ular 36 ventricular) 14 3.16 Footpad 37 3.3 Intradermal 16 3.17 Uncommonroutes 38 3.4 Intramuscular 16 4Special considerations for wildanimals 38 3.5 Intranasal 18 References 39 Correspo ndenceandre q uestsfo rreprintst o:ProfessorD.B.Morton,DepartmentofBiomedicalSciences & BiomedicalEthic s, TheMed icalScho ol,UniversityofBirm ingham,Ed gbaston,Birm inghamB152TT, UK Accepted 14July 2000 # LaboratoryAnimals Ltd. -

Upper and Lower Limb Blocks in Children
Upper and lower limb blocks in children Adrian Bosenberg Correspondence Email: [email protected] INTRODUCTION insulated needles are the next best option Regional anaesthesia is commonly used as an although successful blocks can be achieved with 6 rinciples of clinical anaesthesia adjunct to general anaesthesia and plays a key non-insulated needles. P role in the multimodal approach to perioperative Nerve stimulator 1,2 pain management in children. Peripheral nerve Many peripheral nerve stimulators are available. SUMMARY blockade has increased with the development of Familiarise yourself with a particular device and age-appropriate equipment, safer, long-acting This article outlines regional stick with it. Read the manufacturer’s instructions anaesthetic techniques and local anaesthetic agents, increasing experience and understand how the nerve stimulator works methods used to identify and and low complication rates. before you use it.7 block individual nerves to provide analgesia for surgical The safety of peripheral nerve blocks in children The following basic principles should be followed procedures of the upper has been established in large-scale prospective to locate a peripheral nerve or plexus accurately: and lower limbs. Expertly studies, although caudal epidural remains the performed, peripheral nerve most popular and frequently used block in • Muscle relaxants must be withheld until after blocks can provide long lasting 3-5 completion of the block. anaesthesia and analgesia infants and small children. The experience of for surgery or after injury to the operator, pathology, the site and extent of • Attach the Negative electrode to the Needle the upper or lower limbs in surgery, the child’s body habitus and the presence and the Positive electrode to the Patient using children. -

Drug Delivery Products
INDUSTRY MARKET RESEARCH FOR BUSINESS LEADERS, STRATEGISTS, DECISION MAKERS CLICK TO VIEW Table of Contents 2 List of Tables & Charts 3 Study Overview 4 Sample Text, Table & Chart 5 Sample Profile, Table & Forecast 6 Order Form & Corporate Use License 7 About Freedonia, Custom Research, Related Studies, 8 Drug Delivery Products US Industry Study with Forecasts for 2015 & 2020 Study #2829 | January 2012 | $4800 | 337 pages The Freedonia Group 767 Beta Drive www.freedoniagroup.com Cleveland, OH • 44143-2326 • USA Toll Free US Tel: 800.927.5900 or +1 440.684.9600 Fax: +1 440.646.0484 E-mail: [email protected] Study #2829 January 2012 Drug Delivery Products $4800 337 Pages US Industry Study with Forecasts for 2015 & 2020 Table of Contents Starch Compounds ...................................62 Infusion Pumps ..................................... 150 Cyclodextrins .......................................62 Other Infusion Products .......................... 152 Dextrates ............................................62 Enteral Feeding Supplies ..................... 152 EXECUTIVE SUMMARY Dextrin ...............................................63 IV Accessories ................................... 153 Maltodextrin ........................................63 Parenteral Drug Delivery Devices .................. 154 MARKET ENVIRONMENT Sugars & Polyols ......................................63 Prefillable Syringes ................................ 155 Sucrose ...............................................63 Injectors .......................................... -

Maxillo, Pain EYE & ENT DNB Q & A
Dr. Azam’s Notes in Anesthesiology Postgraduates appearing 3rd Edition for MD, DNB & DA Exams Maxfax, Pain, Eye, ENT & Orthopedic Edited by: Dr. Azam Consultant Anesthesiologist & Critical Care Specialist www.drazam.com 2 Dr Azam’s Notes in Anesthesiology 2013 Dedication To Mohammed Shafiulla, my father, my oxygen, companion, and best friend; for being my major pillar of support and making this vision a reality. Thank you for your continual sacrifices with boundless love and limitless gratitude, for the sake of your children. I owe you a debt I can never repay. I also would like to thank my mom (Naaz Shafi), my wife (Roohi Azam), my two lovely kids (Falaq Zohaa & Mohammed Izaan), for their support, ideas, patience, and encouragement during the many hours of writing this book. Finally, I would like to thank my teachers (Dr.Manjunath Jajoor & team) & Dr T. A. Patil . The dream begins with a teacher who believes in you, who tugs and pushes and leads you to the next plateau, sometimes poking you with a sharp stick called "truth." 3 Dr Azam’s Notes in Anesthesiology 2013 Dr Azam’s Notes in Anesthesiology 2013 A NOTE TO THE READER Anesthesiology is an ever-changing field. Standard safety precautions must be followed, but as new research and clinical experience broaden our knowledge, changes in treatment and drug therapy may become necessary or appropriate. Readers are advised to check the most current product information provided by the manufacturer of each drug to be administered to verify the recommended dose, the method and duration of administration, and contraindications. -

2020 AAHA Anesthesia and Monitoring Guidelines for Dogs and Cats*
VETERINARY PRACTICE GUIDELINES 2020 AAHA Anesthesia and Monitoring Guidelines for Dogs and Cats* Tamara Grubb, DVM, PhD, DACVAAy, Jennifer Sager, BS, CVT, VTS (Anesthesia/Analgesia, ECC)y, James S. Gaynor, DVM, MS, DACVAA, DAIPM, CVA, CVPP, Elizabeth Montgomery, DVM, MPH, Judith A. Parker, DVM, DABVP, Heidi Shafford, DVM, PhD, DACVAA, Caitlin Tearney, DVM, DACVAA ABSTRACT Risk for complications and even death is inherent to anesthesia. However, the use of guidelines, checklists, and training can decrease the risk of anesthesia-related adverse events. These tools should be used not only during the time the patient is unconscious but also before and after this phase. The framework for safe anesthesia delivered as a continuum of care from home to hospital and back to home is presented in these guidelines. The critical importance of client commu- nication and staff training have been highlighted. The role of perioperative analgesia, anxiolytics, and proper handling of fractious/fearful/aggressive patients as components of anesthetic safety are stressed. Anesthesia equipment selection and care is detailed. The objective of these guidelines is to make the anesthesia period as safe as possible for dogs and cats while providing a practical framework for delivering anesthesia care. To meet this goal, tables, algorithms, figures, and “tip” boxes with critical information are included in the manuscript and an in-depth online resource center is available at aaha.org/anesthesia. (J Am Anim Hosp Assoc 2020; 56:---–---. DOI 10.5326/JAAHA-MS-7055) AFFILIATIONS Other recommendations are based on practical clinical experience and From Washington State University College of Veterinary Medicine, Pullman, a consensus of expert opinion.