Cmt Thesis Before
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gentaur Products List
Chapter 2 : Gentaur Products List • Rabbit Anti LAMR1 Polyclonal Antibody Cy5 Conjugated Conjugated • Rabbit Anti Podoplanin gp36 Polyclonal Antibody Cy5 • Rabbit Anti LAMR1 CT Polyclonal Antibody Cy5 • Rabbit Anti phospho NFKB p65 Ser536 Polyclonal Conjugated Conjugated Antibody Cy5 Conjugated • Rabbit Anti CHRNA7 Polyclonal Antibody Cy5 Conjugated • Rat Anti IAA Monoclonal Antibody Cy5 Conjugated • Rabbit Anti EV71 VP1 CT Polyclonal Antibody Cy5 • Rabbit Anti Connexin 40 Polyclonal Antibody Cy5 • Rabbit Anti IAA Indole 3 Acetic Acid Polyclonal Antibody Conjugated Conjugated Cy5 Conjugated • Rabbit Anti LHR CGR Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Integrin beta 7 Polyclonal Antibody Cy5 • Rabbit Anti Natrexone Polyclonal Antibody Cy5 Conjugated • Rabbit Anti MMP 20 Polyclonal Antibody Cy5 Conjugated Conjugated • Rabbit Anti Melamine Polyclonal Antibody Cy5 Conjugated • Rabbit Anti BCHE NT Polyclonal Antibody Cy5 Conjugated • Rabbit Anti NAP1 NAP1L1 Polyclonal Antibody Cy5 • Rabbit Anti Acetyl p53 K382 Polyclonal Antibody Cy5 • Rabbit Anti BCHE CT Polyclonal Antibody Cy5 Conjugated Conjugated Conjugated • Rabbit Anti HPV16 E6 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti CCP Polyclonal Antibody Cy5 Conjugated • Rabbit Anti JAK2 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti HPV18 E6 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti HDC Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Microsporidia protien Polyclonal Antibody Cy5 • Rabbit Anti HPV16 E7 Polyclonal Antibody Cy5 Conjugated • Rabbit Anti Neurocan Polyclonal -

Mice Subjected to Ap2-Cre Mediated Ablation of Microsomal Triglyceride Transfer Protein Are Resistant to High Fat Diet Induced Obesity Ahmed Bakillah1,2* and M
Bakillah and Hussain Nutrition & Metabolism (2016) 13:1 DOI 10.1186/s12986-016-0061-6 RESEARCH Open Access Mice subjected to aP2-Cre mediated ablation of microsomal triglyceride transfer protein are resistant to high fat diet induced obesity Ahmed Bakillah1,2* and M. Mahmood Hussain1,2,3 Abstract Background: Microsomal triglyceride transfer protein (MTP) is essential for the assembly of lipoproteins. MTP has been shown on the surface of lipid droplets of adipocytes; however its function in adipose tissue is not well defined. We hypothesized that MTP may play critical role in adipose lipid droplet formation and expansion. Methods: Plasmids mediated overexpression and siRNA mediated knockdown of Mttp gene were performed in 3T3-L1 pre-adipocytes to evaluate the effects of MTP on cell differentiation and triglyceride accumulation. Adipose- specific knockdown of MTP was achieved in mice bybreeding MTP floxed (Mttpfl/fl) mice with aP2-Cre recombinase transgenic mice. Adipose-specific MTP deficient (A-Mttp-/-) mice were fed 60 % high-fat diet (HFD), and the effects of MTP knockdown on body weight, body fat composition, plasma and tissues lipid composition, glucose metabolism, lipogenesis and intestinal absorption was studied. Lipids were measured in total fasting plasma and size fractionated plasma using colorimetric assays. Gene expression was investigated by Real-Time quantitative PCR. All data was assessed using t-test, ANOVA. Results: MTP expression increased during early differentiation in 3T3-L1 cells, and declined later. The increases in MTP expression preceded PPARγ expression. MTP overexpression enhanced lipid droplets formation, and knockdown attenuated cellular lipid accumulation. These studies indicated that MTP positively affects adipogenesis. -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -
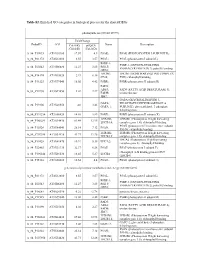
Table S2. Enriched GO Categories in Biological Process for the Shared Degs
Table S2. Enriched GO categories in biological process for the shared DEGs photosynthesis (GO ID:15979) Fold Change ProbeID AGI Col-0(R) pifQ(D) Name Description /Col-0(D) /Col-0(D) A_84_P19035 AT1G30380 17.07 4.9 PSAK; PSAK (PHOTOSYSTEM I SUBUNIT K) A_84_P21372 AT4G12800 8.55 3.57 PSAL; PSAL (photosystem I subunit L) PSBP-1; PSBP-1 (OXYGEN-EVOLVING A_84_P20343 AT1G06680 12.27 3.85 PSII-P; ENHANCER PROTEIN 2); poly(U) binding OEE2; LHCB6; LHCB6 (LIGHT HARVESTING COMPLEX A_84_P14174 AT1G15820 23.9 6.16 CP24; PSII); chlorophyll binding A_84_P11525 AT1G79040 16.02 4.42 PSBR; PSBR (photosystem II subunit R) FAD5; ADS3; FAD5 (FATTY ACID DESATURASE 5); A_84_P19290 AT3G15850 4.02 2.27 FADB; oxidoreductase JB67; GAPA (GLYCERALDEHYDE 3- GAPA; PHOSPHATE DEHYDROGENASE A A_84_P19306 AT3G26650 4.6 3.43 GAPA-1; SUBUNIT); glyceraldehyde-3-phosphate dehydrogenase A_84_P193234 AT2G06520 14.01 3.89 PSBX; PSBX (photosystem II subunit X) LHB1B1; LHB1B1 (Photosystem II light harvesting A_84_P160283 AT2G34430 89.44 32.95 LHCB1.4; complex gene 1.4); chlorophyll binding PSAN (photosystem I reaction center subunit A_84_P10324 AT5G64040 26.14 7.12 PSAN; PSI-N); calmodulin binding LHB1B2; LHB1B2 (Photosystem II light harvesting A_84_P207958 AT2G34420 41.71 12.26 LHCB1.5; complex gene 1.5); chlorophyll binding LHCA2 (Photosystem I light harvesting A_84_P19428 AT3G61470 10.91 5.36 LHCA2; complex gene 2); chlorophyll binding A_84_P22465 AT1G31330 32.37 6.58 PSAF; PSAF (photosystem I subunit F) chlorophyll A-B binding protein CP29 A_84_P190244 AT5G01530 16.45 5.27 LHCB4 -

Fatty Acid Biosynthesis
BI/CH 422/622 ANABOLISM OUTLINE: Photosynthesis Carbon Assimilation – Calvin Cycle Carbohydrate Biosynthesis in Animals Gluconeogenesis Glycogen Synthesis Pentose-Phosphate Pathway Regulation of Carbohydrate Metabolism Anaplerotic reactions Biosynthesis of Fatty Acids and Lipids Fatty Acids contrasts Diversification of fatty acids location & transport Eicosanoids Synthesis Prostaglandins and Thromboxane acetyl-CoA carboxylase Triacylglycerides fatty acid synthase ACP priming Membrane lipids 4 steps Glycerophospholipids Control of fatty acid metabolism Sphingolipids Isoprene lipids: Cholesterol ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1 ANABOLISM II: Biosynthesis of Fatty Acids & Lipids 1. Biosynthesis of fatty acids 2. Regulation of fatty acid degradation and synthesis 3. Assembly of fatty acids into triacylglycerol and phospholipids 4. Metabolism of isoprenes a. Ketone bodies and Isoprene biosynthesis b. Isoprene polymerization i. Cholesterol ii. Steroids & other molecules iii. Regulation iv. Role of cholesterol in human disease ANABOLISM II: Biosynthesis of Fatty Acids & Lipids Lipid Fat Biosynthesis Catabolism Fatty Acid Fatty Acid Degradation Synthesis Ketone body Isoprene Utilization Biosynthesis 2 Catabolism Fatty Acid Biosynthesis Anabolism • Contrast with Sugars – Lipids have have hydro-carbons not carbo-hydrates – more reduced=more energy – Long-term storage vs short-term storage – Lipids are essential for structure in ALL organisms: membrane phospholipids • Catabolism of fatty acids –produces acetyl-CoA –produces reducing -

Saturated Long-Chain Fatty Acid-Producing Bacteria Contribute
Zhao et al. Microbiome (2018) 6:107 https://doi.org/10.1186/s40168-018-0492-6 RESEARCH Open Access Saturated long-chain fatty acid-producing bacteria contribute to enhanced colonic motility in rats Ling Zhao1†, Yufen Huang2†, Lin Lu1†, Wei Yang1, Tao Huang1, Zesi Lin3, Chengyuan Lin1,4, Hiuyee Kwan1, Hoi Leong Xavier Wong1, Yang Chen5, Silong Sun2, Xuefeng Xie2, Xiaodong Fang2,5, Huanming Yang6, Jian Wang6, Lixin Zhu7* and Zhaoxiang Bian1* Abstract Background: The gut microbiota is closely associated with gastrointestinal (GI) motility disorder, but the mechanism(s) by which bacteria interact with and affect host GI motility remains unclear. In this study, through using metabolomic and metagenomic analyses, an animal model of neonatal maternal separation (NMS) characterized by accelerated colonic motility and gut dysbiosis was used to investigate the mechanism underlying microbiota-driven motility dysfunction. Results: An excess of intracolonic saturated long-chain fatty acids (SLCFAs) was associated with enhanced bowel motility in NMS rats. Heptadecanoic acid (C17:0) and stearic acid (C18:0), as the most abundant odd- and even- numbered carbon SLCFAs in the colon lumen, can promote rat colonic muscle contraction and increase stool frequency. Increase of SLCFAs was positively correlated with elevated abundances of Prevotella, Lactobacillus, and Alistipes. Functional annotation found that the level of bacterial LCFA biosynthesis was highly enriched in NMS group. Essential synthetic genes Fabs were largely identified from the genera Prevotella, Lactobacillus, and Alistipes. Pseudo germ-free (GF) rats receiving fecal microbiota from NMS donors exhibited increased defecation frequency and upregulated bacterial production of intracolonic SLCFAs. Modulation of gut dysbiosis by neomycin effectively attenuated GI motility and reduced bacterial SLCFA generation in the colon lumen of NMS rats. -

Crystal Structure of Fabz-ACP Complex Reveals a Dynamic Seesaw-Like Catalytic Mechanism of Dehydratase in Fatty Acid Biosynthesis
Cell Research (2016) 26:1330-1344. ORIGINAL ARTICLE www.nature.com/cr Crystal structure of FabZ-ACP complex reveals a dynamic seesaw-like catalytic mechanism of dehydratase in fatty acid biosynthesis Lin Zhang1, 2, Jianfeng Xiao3, Jianrong Xu1, Tianran Fu1, Zhiwei Cao1, Liang Zhu1, 2, Hong-Zhuan Chen1, 2, Xu Shen3, Hualiang Jiang3, Liang Zhang1, 2 1Department of Pharmacology and Chemical Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, China; 2Shanghai Universities Collaborative Innovation Center for Translational Medicine, Shanghai, China; 3State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China Fatty acid biosynthesis (FAS) is a vital process in cells. Fatty acids are essential for cell assembly and cellular me- tabolism. Abnormal FAS directly correlates with cell growth delay and human diseases, such as metabolic syndromes and various cancers. The FAS system utilizes an acyl carrier protein (ACP) as a transporter to stabilize and shuttle the growing fatty acid chain throughout enzymatic modules for stepwise catalysis. Studying the interactions between enzymatic modules and ACP is, therefore, critical for understanding the biological function of the FAS system. How- ever, the information remains unclear due to the high flexibility of ACP and its weak interaction with enzymatic mod- ules. We present here a 2.55 Å crystal structure of type II FAS dehydratase FabZ in complex with holo-ACP, which exhibits a highly symmetrical FabZ hexamer-ACP3 stoichiometry with each ACP binding to a FabZ dimer subunit. Further structural analysis, together with biophysical and computational results, reveals a novel dynamic seesaw-like ACP binding and catalysis mechanism for the dehydratase module in the FAS system, which is regulated by a critical gatekeeper residue (Tyr100 in FabZ) that manipulates the movements of the β-sheet layer. -

Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals
animals Review Key Genes Regulating Skeletal Muscle Development and Growth in Farm Animals Mohammadreza Mohammadabadi 1 , Farhad Bordbar 1,* , Just Jensen 2 , Min Du 3 and Wei Guo 4 1 Department of Animal Science, Faculty of Agriculture, Shahid Bahonar University of Kerman, Kerman 77951, Iran; [email protected] 2 Center for Quantitative Genetics and Genomics, Aarhus University, 8210 Aarhus, Denmark; [email protected] 3 Washington Center for Muscle Biology, Department of Animal Sciences, Washington State University, Pullman, WA 99163, USA; [email protected] 4 Muscle Biology and Animal Biologics, Animal and Dairy Science, University of Wisconsin-Madison, Madison, WI 53558, USA; [email protected] * Correspondence: [email protected] Simple Summary: Skeletal muscle mass is an important economic trait, and muscle development and growth is a crucial factor to supply enough meat for human consumption. Thus, understanding (candidate) genes regulating skeletal muscle development is crucial for understanding molecular genetic regulation of muscle growth and can be benefit the meat industry toward the goal of in- creasing meat yields. During the past years, significant progress has been made for understanding these mechanisms, and thus, we decided to write a comprehensive review covering regulators and (candidate) genes crucial for muscle development and growth in farm animals. Detection of these genes and factors increases our understanding of muscle growth and development and is a great help for breeders to satisfy demands for meat production on a global scale. Citation: Mohammadabadi, M.; Abstract: Farm-animal species play crucial roles in satisfying demands for meat on a global scale, Bordbar, F.; Jensen, J.; Du, M.; Guo, W. -

Original Article
Original Article Deletion of Rictor in Brain and Fat Alters Peripheral Clock Gene Expression and Increases Blood Pressure Katja Drägert, Indranil Bhattacharya, Giovanni Pellegrini, Petra Seebeck, Abdelhalim Azzi, Steven A. Brown, Stavroula Georgiopoulou, Ulrike Held, Przemyslaw Blyszczuk, Margarete Arras, Rok Humar, Michael N. Hall, Edouard Battegay, Elvira Haas Abstract—The mammalian target of rapamycin complex 2 (mTORC2) contains the essential protein RICTOR and is activated by growth factors. mTORC2 in adipose tissue contributes to the regulation of glucose and lipid metabolism. In the perivascular adipose tissue, mTORC2 ensures normal vascular reactivity by controlling expression of inflammatory molecules. To assess whether RICTOR/mTORC2 contributes to blood pressure regulation, we applied a radiotelemetry approach in control and Rictor knockout (RictoraP2KO) mice generated using adipocyte protein-2 gene promoter–driven CRE recombinase expression to delete Rictor. The 24-hour mean arterial pressure was increased in RictoraP2KO mice, and the physiological decline in mean arterial pressure during the dark period was impaired. In parallel, heart rate and locomotor activity were elevated during the dark period with a pattern similar to blood pressure changes. This phenotype was associated with mild cardiomyocyte hypertrophy, decreased cardiac natriuretic peptides, and their receptor expression in adipocytes. Moreover, clock gene expression was reduced or phase-shifted in perivascular adipose tissue. No differences in clock gene expression were observed in the master clock suprachiasmatic nucleus, although Rictor gene expression was also lower in brain of RictoraP2KO mice. Thus, this study highlights the importance of RICTOR/mTORC2 for interactions between vasculature, adipocytes, and brain to tune physiological outcomes, such as blood pressure and locomotor activity. -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Data-Driven Insights Into Ligands, Proteins, and Genetic Mutations
Data-Driven Insights into Ligands, Proteins, and Genetic Mutations by Jing Lu A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in the University of Michigan 2016 Doctoral Committee: Professor Heather A. Carlson, Chair Professor Charles L. Brooks III Assistant Professor Barry Grant Professor David S. Sept Professor Kerby A. Shedden © Jing Lu, 2016 Acknowledgements I would like to thank my advisor, Dr. Heather Carlson, for years of patient guidance, teaching, and support through the course of my PhD. I have learnt how to think critically and be rigorous in every step of research. I also want to express gratitude to my committee: Professor Charles L. Brooks III, Assistant Professor Barry Grant, Professor David S. Sept, Professor Kerby A. Shedden. Their advising is insightful and deepens my understanding of my research projects. I would like to thank Dr. Richard Smith for timely support for both my writing and research. For many Saturdays and Sundays, he promptly responds my requests for proofreading. Much of my work is built on his code in protein and ligand analysis. I would like to thank other members in Dr. Carlson’s lab for helping me with my work. Through the discussion with Dr. Jim Dunbar, I have learnt many critical ideas in Cheminformatics. Also, thank you to Sarah Graham and Jordan Clark for their tremendous friendship and willing to help with my writing. I would also thank previous members in Dr. Carlson’s lab. I would thank Dr. Phani Ghanakota for many late-night discussions and Dr. -

Adenovirus-Mediated Interference of FABP4 Regulates Mrna Expression of ADIPOQ, LEP and LEPR in Bovine Adipocytes
Adenovirus-mediated interference of FABP4 regulates mRNA expression of ADIPOQ, LEP and LEPR in bovine adipocytes S. Wei1,3, L.S. Zan1,2, H.B. Wang1, G. Cheng1, M. Du3, Z. Jiang3, G.J. Hausman4, D.C. McFarland5 and M.V. Dodson3 1College of Animal Science and Technology, Northwest A&F University, Yangling, Shaanxi, P.R. China 2National Beef Cattle Improvement Center, Yangling, Shaanxi, P.R. China 3Department of Animal Sciences, Washington State University, Pullman, WA, USA 4United States Department of Agriculture, Agriculture Research Services, Athens, GA, USA 5Department of Animal Science, South Dakota State University, Brookings, SD, USA Corresponding authors: L.S. Zan / M.V. Dodson E-mail: [email protected] / [email protected] Genet. Mol. Res. 12 (1): 494-505 (2013) Received May 9, 2012 Accepted September 13, 2012 Published January 4, 2013 DOI http://dx.doi.org/10.4238/2013.January.4.21 ABSTRACT. Fatty acid binding protein 4 (FABP4) is an important adipocyte gene, with roles in fatty acid transport and fat deposition in animals as well as human metabolic syndrome. However, little is known about the functional regulation of FABP4 at the cellular level in bovine. We designed and selected an effective shRNA (small hairpin RNA) against bovine FABP4, constructed a corresponding adenovirus (AD-FABP4), and then detected its influence on mRNA expression of four differentiation-related genes (PPARᵧ, CEBPA, CEBPB, and SREBF1) and three lipid metabolism-related genes (ADIPOQ, LEP and LEPR) of adipocytes. The FABP4 mRNA content, derived from bovine adipocytes, decreased by 41% (P < 0.01) after 24 h and 66% (P < 0.01) Genetics and Molecular Research 12 (1): 494-505 (2013) ©FUNPEC-RP www.funpecrp.com.br shRNA interference of FABP4 in bovine adipocytes 495 after 72 h of AD-FABP4 infection.