Downloaded the Genome Sequences from the Ity Threshold (Input Parameter) Which We Set to 0.6
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Diversity of Understudied Archaeal and Bacterial Populations of Yellowstone National Park: from Genes to Genomes Daniel Colman
University of New Mexico UNM Digital Repository Biology ETDs Electronic Theses and Dissertations 7-1-2015 Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes Daniel Colman Follow this and additional works at: https://digitalrepository.unm.edu/biol_etds Recommended Citation Colman, Daniel. "Diversity of understudied archaeal and bacterial populations of Yellowstone National Park: from genes to genomes." (2015). https://digitalrepository.unm.edu/biol_etds/18 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in Biology ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Daniel Robert Colman Candidate Biology Department This dissertation is approved, and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: Cristina Takacs-Vesbach , Chairperson Robert Sinsabaugh Laura Crossey Diana Northup i Diversity of understudied archaeal and bacterial populations from Yellowstone National Park: from genes to genomes by Daniel Robert Colman B.S. Biology, University of New Mexico, 2009 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy Biology The University of New Mexico Albuquerque, New Mexico July 2015 ii DEDICATION I would like to dedicate this dissertation to my late grandfather, Kenneth Leo Colman, associate professor of Animal Science in the Wool laboratory at Montana State University, who even very near the end of his earthly tenure, thought it pertinent to quiz my knowledge of oxidized nitrogen compounds. He was a man of great curiosity about the natural world, and to whom I owe an acknowledgement for his legacy of intellectual (and actual) wanderlust. -

Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline
Phylogenetics of Archaeal Lipids Amy Kelly 9/27/2006 Outline • Phlogenetics of Archaea • Phlogenetics of archaeal lipids • Papers Phyla • Two? main phyla – Euryarchaeota • Methanogens • Extreme halophiles • Extreme thermophiles • Sulfate-reducing – Crenarchaeota • Extreme thermophiles – Korarchaeota? • Hyperthermophiles • indicated only by environmental DNA sequences – Nanoarchaeum? • N. equitans a fast evolving euryarchaeal lineage, not novel, early diverging archaeal phylum – Ancient archael group? • In deepest brances of Crenarchaea? Euryarchaea? Archaeal Lipids • Methanogens – Di- and tetra-ethers of glycerol and isoprenoid alcohols – Core mostly archaeol or caldarchaeol – Core sometimes sn-2- or Images removed due to sn-3-hydroxyarchaeol or copyright considerations. macrocyclic archaeol –PMI • Halophiles – Similar to methanogens – Exclusively synthesize bacterioruberin • Marine Crenarchaea Depositional Archaeal Lipids Biological Origin Environment Crocetane methanotrophs? methane seeps? methanogens, PMI (2,6,10,15,19-pentamethylicosane) methanotrophs hypersaline, anoxic Squalane hypersaline? C31-C40 head-to-head isoprenoids Smit & Mushegian • “Lost” enzymes of MVA pathway must exist – Phosphomevalonate kinase (PMK) – Diphosphomevalonate decarboxylase – Isopentenyl diphosphate isomerase (IPPI) Kaneda et al. 2001 Rohdich et al. 2001 Boucher et al. • Isoprenoid biosynthesis of archaea evolved through a combination of processes – Co-option of ancestral enzymes – Modification of enzymatic specificity – Orthologous and non-orthologous gene -

A Late Methanogen Origin for Molybdenum-Dependent Nitrogenase E
Geobiology (2011), 9, 221–232 DOI: 10.1111/j.1472-4669.2011.00278.x A late methanogen origin for molybdenum-dependent nitrogenase E. S. BOYD,1 A. D. ANBAR,2 S. MILLER,3 T. L. HAMILTON,1 M. LAVIN4 ANDJ.W.PETERS1 1Department of Chemistry and Biochemistry and the Astrobiology Biogeocatalysis Research Center, Montana State University, Bozeman, MT, USA 2School of Earth and Space Exploration and Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ, USA 3Department of Biological Sciences, University of Montana, Missoula, MT, USA 4Department of Plant Sciences, Montana State University, Bozeman, MT, USA ABSTRACT Mounting evidence indicates the presence of a near complete biological nitrogen cycle in redox-stratified oceans during the late Archean to early Proterozoic (c. 2.5–2.0 Ga). It has been suggested that the iron (Fe)- or vana- dium (V)-dependent nitrogenase rather than molybdenum (Mo)-dependent form was responsible for dinitrogen fixation during this time because oceans were depleted in Mo and rich in Fe. We evaluated this hypothesis by examining the phylogenetic relationships of proteins that are required for the biosynthesis of the active site cofactor of Mo-nitrogenase in relation to structural proteins required for Fe-, V- and Mo-nitrogenase. The results are highly suggestive that among extant nitrogen-fixing organisms for which genomic information exists, Mo-nitrogenase is unlikely to have been associated with the Last Universal Common Ancestor. Rather, the origin of Mo-nitrogenase can be traced to an ancestor of the anaerobic and hydrogenotrophic methanogens with acquisition in the bacterial domain via lateral gene transfer involving an anaerobic member of the Firmicutes.A comparison of substitution rates estimated for proteins required for the biosynthesis of the nitrogenase active site cofactor and for a set of paralogous proteins required for the biosynthesis of bacteriochlorophyll suggests that Nif emerged from a nitrogenase-like ancestor approximately 1.5–2.2 Ga. -

Discovery of a Novel Methanogen Prevalent in Thawing Permafrost
ARTICLE Received 11 Jun 2013 | Accepted 7 Jan 2014 | Published 14 Feb 2014 DOI: 10.1038/ncomms4212 Discovery of a novel methanogen prevalent in thawing permafrost Rhiannon Mondav1,*,w, Ben J. Woodcroft1,*, Eun-Hae Kim2, Carmody K. McCalley3,w, Suzanne B. Hodgkins4, Patrick M. Crill5, Jeffrey Chanton4, Gregory B. Hurst6, Nathan C. VerBerkmoes6,w, Scott R. Saleska3, Philip Hugenholtz1, Virginia I. Rich2 & Gene W. Tyson1 Thawing permafrost promotes microbial degradation of cryo-sequestered and new carbon leading to the biogenic production of methane, creating a positive feedback to climate change. Here we determine microbial community composition along a permafrost thaw gradient in northern Sweden. Partially thawed sites were frequently dominated by a single archaeal phylotype, Candidatus ‘Methanoflorens stordalenmirensis’ gen. nov. sp. nov., belonging to the uncultivated lineage ‘Rice Cluster II’ (Candidatus ‘Methanoflorentaceae’ fam. nov.). Metage- nomic sequencing led to the recovery of its near-complete genome, revealing the genes necessary for hydrogenotrophic methanogenesis. These genes are highly expressed and methane carbon isotope data are consistent with hydrogenotrophic production of methane in the partially thawed site. In addition to permafrost wetlands, ‘Methanoflorentaceae’ are widespread in high methane-flux habitats suggesting that this lineage is both prevalent and a major contributor to global methane production. In thawing permafrost, Candidatus ‘M. stordalenmirensis’ appears to be a key mediator of methane-based positive feedback to climate warming. 1 Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane 4072, Queensland, Australia. 2 Department of Soil, Water and Environmental Science, University of Arizona, Tucson, Arizona 85721, USA. 3 Ecology and Evolutionary Biology Department, University of Arizona, Tucson, Arizona 85721, USA. -

Archaeology of Eukaryotic DNA Replication
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Archaeology of Eukaryotic DNA Replication Kira S. Makarova and Eugene V. Koonin National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland 20894 Correspondence: [email protected] Recent advances in the characterization of the archaeal DNA replication system together with comparative genomic analysis have led to the identification of several previously un- characterized archaeal proteins involved in replication and currently reveal a nearly com- plete correspondence between the components of the archaeal and eukaryotic replication machineries. It can be inferred that the archaeal ancestor of eukaryotes and even the last common ancestor of all extant archaea possessed replication machineries that were compa- rable in complexity to the eukaryotic replication system. The eukaryotic replication system encompasses multiple paralogs of ancestral components such that heteromeric complexes in eukaryotes replace archaeal homomeric complexes, apparently along with subfunctionali- zation of the eukaryotic complex subunits. In the archaea, parallel, lineage-specific dupli- cations of many genes encoding replication machinery components are detectable as well; most of these archaeal paralogs remain to be functionally characterized. The archaeal rep- lication system shows remarkable plasticity whereby even some essential components such as DNA polymerase and single-stranded DNA-binding protein are displaced by unrelated proteins with analogous activities in some lineages. ouble-stranded DNA is the molecule that Okazaki fragments (Kornberg and Baker 2005; Dcarries genetic information in all cellular Barry and Bell 2006; Hamdan and Richardson life-forms; thus, replication of this genetic ma- 2009; Hamdan and van Oijen 2010). -

Yu-Chen Ling and John W. Moreau
Microbial Distribution and Activity in a Coastal Acid Sulfate Soil System Introduction: Bioremediation in Yu-Chen Ling and John W. Moreau coastal acid sulfate soil systems Method A Coastal acid sulfate soil (CASS) systems were School of Earth Sciences, University of Melbourne, Melbourne, VIC 3010, Australia formed when people drained the coastal area Microbial distribution controlled by environmental parameters Microbial activity showed two patterns exposing the soil to the air. Drainage makes iron Microbial structures can be grouped into three zones based on the highest similarity between samples (Fig. 4). Abundant populations, such as Deltaproteobacteria, kept constant activity across tidal cycling, whereas rare sulfides oxidize and release acidity to the These three zones were consistent with their geological background (Fig. 5). Zone 1: Organic horizon, had the populations changed activity response to environmental variations. Activity = cDNA/DNA environment, low pH pore water further dissolved lowest pH value. Zone 2: surface tidal zone, was influenced the most by tidal activity. Zone 3: Sulfuric zone, Abundant populations: the heavy metals. The acidity and toxic metals then Method A Deltaproteobacteria Deltaproteobacteria this area got neutralized the most. contaminate coastal and nearby ecosystems and Method B 1.5 cause environmental problems, such as fish kills, 1.5 decreased rice yields, release of greenhouse gases, Chloroflexi and construction damage. In Australia, there is Gammaproteobacteria Gammaproteobacteria about a $10 billion “legacy” from acid sulfate soils, Chloroflexi even though Australia is only occupied by around 1.0 1.0 Cyanobacteria,@ Acidobacteria Acidobacteria Alphaproteobacteria 18% of the global acid sulfate soils. Chloroplast Zetaproteobacteria Rare populations: Alphaproteobacteria Method A log(RNA(%)+1) Zetaproteobacteria log(RNA(%)+1) Method C Method B 0.5 0.5 Cyanobacteria,@ Bacteroidetes Chloroplast Firmicutes Firmicutes Bacteroidetes Planctomycetes Planctomycetes Ac8nobacteria Fig. -
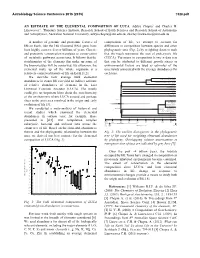
An Estimate of the Elemental Composition of Luca
Astrobiology Science Conference 2015 (2015) 7328.pdf AN ESTIMATE OF THE ELEMENTAL COMPOSITION OF LUCA. Aditya Chopra1 and Charles H. Lineweaver1, 1Planetary Science Institute, Research School of Earth Sciences and Research School of Astronomy and Astrophysics, Australian National University, [email protected], [email protected] A number of genomic and proteomic features of composition of life, we attempt to account for life on Earth, like the 16S ribosomal RNA gene, have differences in composition between species and other been highly conserved over billions of years. Genetic phylogenetic taxa (Fig. 2) by weighting datasets such and proteomic conservation translates to conservation that the result represents the root of prokaryotic life of metabolic pathways across taxa. It follows that the (LUCA). Variations in composition between data sets stoichiometry of the elements that make up some of that can be attributed to different growth stages or the biomolecules will be conserved. By extension, the environmental factors are used as estimates of the elemental make up of the whole organism is a uncertainty associated with the average abundances for relatively conserved feature of life on Earth [1,2]. each taxa. We describe how average bulk elemental Euryarchaeota 1a Methanococci, Methanobacteria, Methanopyri 7 Euryarchaeota 1b abundances in extant life can yield an indirect estimate 6 Thermoplasmata, Methanomicrobia, Halobacteria, Archaeoglobi Euryarchaeota 2 4 Thermococci of relative abundances of elements in the Last Crenarchaeota Sulfolobus, Thermoproteus ? Thaumarchaeota Universal Common Ancestor (LUCA). The results Cenarchaeum ? Korarchaeota ARMAN could give us important hints about the stoichiometry ? 2 Archaeal Richmond Mine Acidophilic Nanoorganisms Nanoarchaeota of the environment where LUCA existed and perhaps Archaea Terrabacteria clues to the processes involved in the origin and early Actinobacteria, Deinococcus-Thermus, 1 Cyanobacteria, Life Chloroflexi, 9 evolution of life [3]. -

METHANOGENS AS MODELS for LIFE on MARS. R. L. Mickol1, W. H. Waddell2, and T
Eighth International Conference on Mars (2014) 1005.pdf METHANOGENS AS MODELS FOR LIFE ON MARS. R. L. Mickol1, W. H. Waddell2, and T. A. Kral1,3, 1Arkansas Center for Space and Planetary Sciences, 202 Old Museum Building, University of Arkansas, Fayetteville, Arkansas, 72701, USA, [[email protected]], 2Dept. of Health, Human Performance, and Recreation, HPER 308, University of Arkansas, Fayetteville, Arkansas, 72701, USA, 3Dept. of Biological Sciences, SCEN 632, University of Arkansas, Fayetteville, Arkansas, 72701, USA. Introduction: The discovery of methane in the Sciences, University of Arkansas, Fayetteville, Arkan- martian atmosphere [1-4] has fueled the study of meth- sas. All four methanogen species were tested in these anogens as ideal candidates for life on Mars. Methano- experiments. Methanogens were grown in their respec- gens are chemoautotrophs from the domain Archaea. tive anaerobic growth media and placed into the cham- These microorganisms utilize hydrogen as an energy ber with a palladium catalyst box to remove residual source and carbon dioxide as a carbon source to pro- oxygen. The chamber was evacuated to a pre- duce methane. Methanogens can be considered ideal determined pressure and filled with 80:20 H2:CO2 gas. candidates for life on Mars because they are anaerobic, This procedure was repeated three times to ensure re- they do not require organic nutrients and are non- moval of the atmosphere. The chamber was then main- photosynthetic, indicating they could exist in sub- tained at the desired pressure (133-143 mbar, 67-72 surface environments. mbar, 33-38 mbar, 6-10 mbar, 7-20 mbar) for the dura- Our lab has studied methanogens as models for life tion of the experiments. -

Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio
International Journal of New Technology and Research (IJNTR) ISSN:2454-4116, Volume-2, Issue-9, September 2016 Pages 81-86 Establishment and Analysis of Microbial Communities Capable of Producing Methane from Grass Waste at Extremely High C/N Ratio S. Matsuda, T. Ohtsuki* Anaerobic digestion is a conventional method for biomass Abstract— Acclimation of microbial communities, aiming to utilization [8]. Despite the fact that a great deal of research methane production from grass as a sole substrate at extremely for methane production from grass has been conducted high carbon-to-nitrogen (C/N) ratio, was conducted. In a series especially in recent years, almost processes need supply of of experiments with various sizes of added grass, two microbial communities showing high methane production were obtained abundant sludge source such as sewage and manure [9, 10]. with powdered grass. In the two microbial communities Many studies indicated that the optimal carbon-to-nitrogen designated NR and RP, Bacteroidia including genera (C/N) ratio in methane fermentation were 25-30, and the C/N Bacteroides, Dysgonomonas, Proteiniphilum, and Alistipes were ratio higher than 40 are not generally suitable [11]. The C/N detected as dominant members in eubacteria. It was also shown ration of raw grass shows a wide range; wild grassy weed is at that Methanomicrobia and Methanobacteria including genera relatively high (>59) while lawn grass is at low (<29) [12]. Methanomassiliicoccus and Methanobacterium were found as dominant members in methanogen. It is noteworthy that Therefore, it is difficult to conduct methane fermentation nitrogen fixation were observed both in NR and RP, suggesting with wild grass-only, also being due to less degradability of that insufficiency of nitrogen sources would be complemented lignocellulose [13]. -

Methanogenic Burst in the End-Permian Carbon Cycle
Methanogenic burst in the end-Permian carbon cycle Daniel H. Rothmana,b,1, Gregory P. Fournierc, Katherine L. Frenchb, Eric J. Almc, Edward A. Boyleb, Changqun Caod, and Roger E. Summonsb aLorenz Center, bDepartment of Earth, Atmospheric, and Planetary Sciences, and cDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139; and dState Key Laboratory of Palaeobiology and Stratigraphy, Nanjing Institute of Geology and Palaeontology, Chinese Academy of Sciences, Nanjing 210008, China Edited by John M. Hayes, Woods Hole Oceanographic Institution, Woods Hole, MA, and approved February 4, 2014 (received for review September 27, 2013) The end-Permian extinction is associated with a mysterious disrup- and its perturbation to the carbon cycle but also to amplify the tion to Earth’s carbon cycle. Here we identify causal mechanisms via development of marine anoxia. Taken as a whole, these results three observations. First, we show that geochemical signals indicate reconcile an array of apparently disparate observations about the superexponential growth of the marine inorganic carbon reservoir, end-Permian event. coincident with the extinction and consistent with the expansion of a new microbial metabolic pathway. Second, we show that the effi- Growth of the Marine Carbon Reservoir cient acetoclastic pathway in Methanosarcina emerged at a time sta- Fig. 1A displays the carbon-isotopic record in Meishan, China tistically indistinguishable from the extinction. Finally, we show that (5). Immediately preceding the extinction event, the isotopic com- nickel concentrations in South China sediments increased sharply at position δ1 of carbonate carbon declines by about 7‰ during the extinction, probably as a consequence of massive Siberian volca- a period of about 100 thousand years (Kyr), first slowly, and sub- nism, enabling a methanogenic expansion by removal of nickel lim- sequently rapidly. -

Supplementary Information
Retroconversion of estrogens into androgens by bacteria via a cobalamin-mediated methylation Po-Hsiang Wang, Yi-Lung Chen, Sean Ting-Shyang Wei, Kan Wu, Tzong-Huei Lee, Tien-Yu Wu, and Yin-Ru Chiang Supplementary Information Table of Contents Dataset Dataset S1. Genome annotation of strain DHT3 and transcriptomic analysis (RNA-Seq) of bacterial cells grown anaerobically with testosterone or estradiol. SI Tables Table S1. Oligonucleotides used in this study. Table S2. Selection of housekeeping genes of strain DHT3 used for constructing the linear regression line in the global gene expression profiles (RNA-Seq). Table S3. Selection of the cobalamin-dependent methyltransferases used for the un-rooted maximum likelihood tree construction. Table S4. UPLC–APCI–HRMS data of the intermediates involved in anaerobic estrone catabolism by strain DHT3. Table S5. 1H- (600 MHz) and 13C-NMR (150 MHz) spectral data of the HPLC-purified metabolite (AND2) and the authentic standard 5-androstan-3,17-diol Table S6. Selection of the bacteria used for comparative analysis of the gene organization for HIP degradation. SI Figures Fig. S1 Scanning electron micrographs of strain DHT3 cells. Fig. S2 Cobalamin as an essential vitamin during the anaerobic growth of strain DHT3 on estradiol. Fig. S3 Arrangement and expression analysis of the emt genes in strain DHT3. Fig. S4 The anaerobic growth of the wild type (A) and the emtA-disrupted mutant (B) of strain DHT3 with testosterone and estradiol. Fig. S5 APCI–HRMS spectrum of the HIP produced by estrone-fed strain DHT3. 1 Fig. S6 UPLC–APCI–HRMS spectra of two TLC-purified androgen metabolites, 17β-hydroxyandrostan-3-one (A) and 3β,17β-dihydroxyandrostane (B). -

Universidad Politécnica De Madrid Escuela Técnica Superior De Ingeniería Agronómica Alimentaria Y De Biosistemas Departamento De Biotecnología-Biología Vegetal
UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL TESIS DOCTORAL Caracterización de una nueva proteína hipertermófila NifB y su papel en el mecanismo de síntesis de NifB-co, precursor del grupo metálico de FeMo-co Author: Alessandro Scandurra Director: Prof. Luis Manuel Rubio Herrero Co-director: Dr. Simon Jean Marius Arragain i ii UNIVERSIDAD POLITÉCNICA DE MADRID ESCUELA TÉCNICA SUPERIOR DE INGENIERÍA AGRONÓMICA ALIMENTARIA Y DE BIOSISTEMAS DEPARTAMENTO DE BIOTECNOLOGÍA-BIOLOGÍA VEGETAL Memoria presentada por D. Alessandro Scandurra para optar al grado de Doctor Director Dr. Luis Manuel Rubio Herrero Profesor Titular UPM Madrid, 2017 © This copy oF the thesis has been supplied on condition that anyone who consults it is understood to recognize that its copyright rest with the author and that no quotation From the thesis, or any inFormation derived therefrom may be published without the author’s prior, written consent. I II AcKnowledgments As each journey, also this one has an end (Thanks goodness). It was a long trip, with several changes oF direction. Like For many other people, the PhD adventure was not easy, but I have to admit that I was lucky enough to meet really unique people, that became special Friendships. First oF all, I want to thank my Family, especially my mother and my Father, who always push me to go Forward in my life and to Follow my dreams, ready to pay the sacrifice oF the distance. This expirience has helped me to understand how easy is to be a son and how challenging is to be a parent.