Method 625.1: Base/Neutrals and Acids by GC/MS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

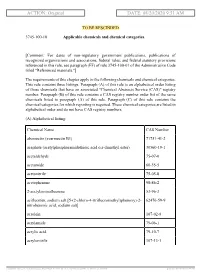
3745-100-10 Applicable Chemicals and Chemical Categories
3745-100-10 Applicable chemicals and chemical categories. [Comment: For dates of non-regulatory government publications, publications of recognized organizations and associations, federal rules, and federal statutory provisions referenced in this rule, see the "Incorporation by Reference" section at the end of rule 3745-100-01.] The requirements of this chapter apply to the following chemicals and chemical categories. This rule contains three listings. Paragraph (A) of this rule is an alphabetical order listing of those chemicals that have an associated "Chemical Abstracts Service (CAS)" registry number. Paragraph (B) of this rule contains a CAS registry number order list of the same chemicals listed in paragraph (A) of this rule. Paragraph (C) of this rule contains the chemical categories for which reporting is required. These chemical categories are listed in alphabetical order and do not have CAS registry numbers. (A) Alphabetical listing: -- Chemical Name CAS Number abamectin (avermectin B1) 71751-41-2 acephate (acetylphosphoramidothioic acid o,s-dimethyl ester) 30560-19-1 acetaldehyde 75-07-0 acetamide 60-35-5 acetonitrile 75-05-8 acetophenone 98-86-2 2-acetylaminofluorene 53-96-3 acifluorfen, sodium salt (5- (2-chloro-4- (trifluoromethyl) - phenoxy)-2-nitro-benzoic acid, sodium salt) 62476-59-9 acrolein 107-02-8 acrylamide 79-06-1 acrylic acid 79-10-7 acrylonitrile 107-13-1 alachlor 15972-60-8 aldicarb 116-06-3 aldrin [1,4,5,8-dimethanonaphthalene, 1,2,3,4,10,10-hexachloro- 1,4,4A,5,8,8a-hexahydro- (1 alpha, 4 alpha, 4a beta, 5 -

C–H Arylation of Triphenylene, Naphthalene and Related Arenes Using Pd/C† Cite This: Chem
Chemical Science View Article Online EDGE ARTICLE View Journal | View Issue C–H arylation of triphenylene, naphthalene and related arenes using Pd/C† Cite this: Chem. Sci.,2015,6,1816 Karl D. Collins, Roman Honeker,‡ Suhelen Vasquez-C´ espedes,´ ‡ Dan-Tam D. Tang and Frank Glorius* A highly selective arylation of a number of polyaromatic hydrocarbons (PAHs) with aryliodonium salts and Pd/C as the only reagent is reported. The first C–H functionalization of triphenylene is explored, and proceeds at the most sterically hindered position. This non-chelate assisted C–H functionalization Received 4th October 2014 extends the reactivity profile of Pd/C and provides controlled access to p-extended PAHs, an important Accepted 19th December 2014 aspect of work towards the preparation of nanographenes. Mechanistic studies suggest in situ formation DOI: 10.1039/c4sc03051f of catalytically active insoluble nanoparticles, and that the reaction likely proceeds via a Pd(0)/Pd(II) type www.rsc.org/chemicalscience reaction manifold. Creative Commons Attribution 3.0 Unported Licence. Introduction precedent for the direct arylation of larger PAHs is reported. Seminal work by Oi and Inoue reported an effective arylation of Pd/C has long been established as an efficient catalyst in phenanthrene and uoranthene with aryltin trichlorides.10 One hydrogenation and cross-coupling reactions.1 Although sup- example of phenanthrene arylation has also been reported by ported catalysts are formally heterogeneous in nature, studies of Shi.11 Important studies by the group of Itami demonstrated a Pd/C suggest it typically acts as a reservoir of homogeneous more general solution (Scheme 1) using prepared boroxines as active catalytic species, following leaching of palladium from coupling partners.7d 1c,1e,2 the support into the solution. -

Mechanistic Study of Methylbenzene Dealkylation in Methanol-To-Olefins
Journal of Catalysis 369 (2019) 86–94 Contents lists available at ScienceDirect Journal of Catalysis journal homepage: www.elsevier.com/locate/jcat Mechanistic study of methylbenzene dealkylation in methanol-to-olefins catalysis on HSAPO-34 ⇑ Andrew Hwang, Blake A. Johnson, Aditya Bhan Department of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, MN 55455, USA article info abstract Article history: Methylbenzenes entrained within the cavities of silicoaluminophosphate zeotype HSAPO-34 react with Received 14 June 2018 methanol in H+-mediated dealkylation to give ethylene and propylene in methanol-to-olefins catalysis. Revised 1 August 2018 Methylbenzenes dealkylation on solid acids is proposed to occur either via the side-chain mechanism, Accepted 14 October 2018 where an exocyclic C@C undergoes successive methylation prior to CAC cleavage for olefin elimination, or the paring mechanism, where ring contraction to a bicyclohexenyl cation precedes CAC cleavage for olefin elimination. Distinct dealkylation mechanisms prescribe distinct combinations of C atoms—from Keywords: aromatic methyl, aromatic ring, and methanol/dimethyl ether—to comprise the olefin product. Site- Methanol-to-olefins specific isotope tracing that distinguishes between isotope labels in aromatic methyl and aromatic ring Methylbenzenes dealkylation Isotope tracing positions for each methylbenzene shows that tetramethylbenzene gives ethylene via the side-chain Site-specific mechanism and penta- and hexamethylbenzene give propylene via the paring mechanism. The ratio of Quantitative 13C NMR propylene selectivity to ethylene selectivity increases in methanol reactions on HSAPO-34 entrained with HSAPO-34 a distribution of methylbenzenes deliberately manipulated towards increasing fractions of penta- and Methylbenzene flash chromatography hexamethylbenzene, corroborating the conclusion that aromatic precursors and dealkylation mecha- nisms for ethylene diverge from those for propylene. -

Polycyclic Aromatic Hydrocarbon Structure Index
NIST Special Publication 922 Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899-0001 December 1997 revised August 2020 U.S. Department of Commerce William M. Daley, Secretary Technology Administration Gary R. Bachula, Acting Under Secretary for Technology National Institute of Standards and Technology Raymond G. Kammer, Director Polycyclic Aromatic Hydrocarbon Structure Index Lane C. Sander and Stephen A. Wise Chemical Science and Technology Laboratory National Institute of Standards and Technology Gaithersburg, MD 20899 This tabulation is presented as an aid in the identification of the chemical structures of polycyclic aromatic hydrocarbons (PAHs). The Structure Index consists of two parts: (1) a cross index of named PAHs listed in alphabetical order, and (2) chemical structures including ring numbering, name(s), Chemical Abstract Service (CAS) Registry numbers, chemical formulas, molecular weights, and length-to-breadth ratios (L/B) and shape descriptors of PAHs listed in order of increasing molecular weight. Where possible, synonyms (including those employing alternate and/or obsolete naming conventions) have been included. Synonyms used in the Structure Index were compiled from a variety of sources including “Polynuclear Aromatic Hydrocarbons Nomenclature Guide,” by Loening, et al. [1], “Analytical Chemistry of Polycyclic Aromatic Compounds,” by Lee et al. [2], “Calculated Molecular Properties of Polycyclic Aromatic Hydrocarbons,” by Hites and Simonsick [3], “Handbook of Polycyclic Hydrocarbons,” by J. R. Dias [4], “The Ring Index,” by Patterson and Capell [5], “CAS 12th Collective Index,” [6] and “Aldrich Structure Index” [7]. In this publication the IUPAC preferred name is shown in large or bold type. -
ACTION: Original DATE: 08/20/2020 9:51 AM
ACTION: Original DATE: 08/20/2020 9:51 AM TO BE RESCINDED 3745-100-10 Applicable chemicals and chemical categories. [Comment: For dates of non-regulatory government publications, publications of recognized organizations and associations, federal rules, and federal statutory provisions referenced in this rule, see paragraph (FF) of rule 3745-100-01 of the Administrative Code titled "Referenced materials."] The requirements of this chapter apply to the following chemicals and chemical categories. This rule contains three listings. Paragraph (A) of this rule is an alphabetical order listing of those chemicals that have an associated "Chemical Abstracts Service (CAS)" registry number. Paragraph (B) of this rule contains a CAS registry number order list of the same chemicals listed in paragraph (A) of this rule. Paragraph (C) of this rule contains the chemical categories for which reporting is required. These chemical categories are listed in alphabetical order and do not have CAS registry numbers. (A) Alphabetical listing: Chemical Name CAS Number abamectin (avermectin B1) 71751-41-2 acephate (acetylphosphoramidothioic acid o,s-dimethyl ester) 30560-19-1 acetaldehyde 75-07-0 acetamide 60-35-5 acetonitrile 75-05-8 acetophenone 98-86-2 2-acetylaminofluorene 53-96-3 acifluorfen, sodium salt [5-(2-chloro-4-(trifluoromethyl)phenoxy)-2- 62476-59-9 nitrobenzoic acid, sodium salt] acrolein 107-02-8 acrylamide 79-06-1 acrylic acid 79-10-7 acrylonitrile 107-13-1 [ stylesheet: rule.xsl 2.14, authoring tool: RAS XMetaL R2_0F1, (dv: 0, p: 185720, pa: -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

Measurements of Higher Alkanes Using NO Chemical Ionization in PTR-Tof-MS
Atmos. Chem. Phys., 20, 14123–14138, 2020 https://doi.org/10.5194/acp-20-14123-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Measurements of higher alkanes using NOC chemical ionization in PTR-ToF-MS: important contributions of higher alkanes to secondary organic aerosols in China Chaomin Wang1,2, Bin Yuan1,2, Caihong Wu1,2, Sihang Wang1,2, Jipeng Qi1,2, Baolin Wang3, Zelong Wang1,2, Weiwei Hu4, Wei Chen4, Chenshuo Ye5, Wenjie Wang5, Yele Sun6, Chen Wang3, Shan Huang1,2, Wei Song4, Xinming Wang4, Suxia Yang1,2, Shenyang Zhang1,2, Wanyun Xu7, Nan Ma1,2, Zhanyi Zhang1,2, Bin Jiang1,2, Hang Su8, Yafang Cheng8, Xuemei Wang1,2, and Min Shao1,2 1Institute for Environmental and Climate Research, Jinan University, 511443 Guangzhou, China 2Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, 511443 Guangzhou, China 3School of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), 250353 Jinan, China 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Protection and Resources Utilization, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, 510640 Guangzhou, China 5State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, 100871 Beijing, China 6State Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese -

Supplement of Compilation and Evaluation of Gas Phase Diffusion Coefficients of Reactive Trace Gases in the Atmosphere
Supplement of Atmos. Chem. Phys., 15, 5585–5598, 2015 http://www.atmos-chem-phys.net/15/5585/2015/ doi:10.5194/acp-15-5585-2015-supplement © Author(s) 2015. CC Attribution 3.0 License. Supplement of Compilation and evaluation of gas phase diffusion coefficients of reactive trace gases in the atmosphere: Volume 2. Diffusivities of organic compounds, pressure-normalised mean free paths, and average Knudsen numbers for gas uptake calculations M. J. Tang et al. Correspondence to: M. J. Tang ([email protected]) and M. Kalberer ([email protected]) The copyright of individual parts of the supplement might differ from the CC-BY 3.0 licence. Table of Contents 1 Alkanes and cycloalkanes ............................................................................................ 1 1.1 CH 4 (methane), C 2H6 (ethane), and C 3H8 (propane) ............................................. 1 1.2 C 4H10 (butane, methyl propane) ............................................................................ 3 1.3 C 5H12 (n-pentane, methyl butane, dimethyl butane) ............................................. 5 1.4 C 6H14 (n-hexane, 2,3-dimethyl butane) ................................................................ 7 1.5 C 7H16 (n-heptane, 2,4-dimethyl pentane).............................................................. 9 1.6 C 8H18 (n-octane, 2,2,4-trimethyl pentane) .......................................................... 11 1.7 C 9H20 (n-nonane), C 10 H22 (n-decane, 2,3,3-trimethyl heptane) and C 12 H26 (n- dodecane) ................................................................................................................. -

Supporting Information
Electronic Supplementary Material (ESI) for ChemComm. This journal is © The Royal Society of Chemistry 2015 Electronic Supplementary Information for Anodic aromatic C,C cross-coupling reaction using parallel laminar flow mode in a flow microreactor Toshihiro Arai, Hiroyuki Tateno, Koji Nakabayashi, Tsuneo Kashiwagi, and Mahito Atobe Department of Environment and System Sciences, Yokohama National University, 79-7 Tokiwadai, Hodogaya-ku, Yokohama 2408501, Japan. *To whom the correspondence should be addressed. E-mail: [email protected] This PDF file includes: Instrumentation Materials Experimental procedure Spectroscopic data S1 1. Instrumentation Nuclear magnetic resonance (1H NMR) spectra were measured on BRUKER DRX 300 1 1 spectrometer operating at 300 MHz ( H NMR) in CDCl3. All H NMR chemical shifts were reported in ppm relative to internal references of TMS at 0.00. Preparative electrolyses were carried out with a HOKUTO DENKO HABF-501A Potentiostat/Galvanostat. GCMS analyses were performed with a Shimadzu gas chromatograph mass spectrometer GCMS-QP2010. 2. Materials Acetonitrile, acetic acid, and naphthalene (1) were purchased from Kanto Chemical and used as received. Pentamethylbenzene (2), isodurene (5), mesitylene (6), 2-bromonaphthalene (8), and tetrabutylammonium tetrafluoroborate (Bu4NBF4) were purchased from Tokyo Chemical Industry and used as received. 3. Flow Microreactor Figure S1 shows construction procedure for the electrochemical two-inlet flow microreactor. The reactor was constructed from glass plates and two platinum (Pt) plates (3 cm width, 3 cm length each) (Step 1 of Figure S1). A spacer (20 m thickness double faced adhesive tape) was used to leave a rectangular channel exposed, and the two electrodes were simply sandwiched together (area of the two electrodes: 1 × 3 cm2). -
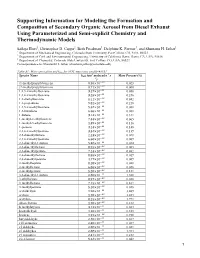
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -

Laboratory Spectroscopy and Astronomical Significance of The
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repository@Nottingham Laboratory spectroscopy and astronomical significance of the fully-benzenoid PAH triphenylene and its cation V. Kofmana,b, P.J. Sarrec, R.E. Hibbinsc,d, I.L. ten Kateb, H. Linnartza aSackler Laboratory for Astrophysics, Leiden Observatory, Leiden University, PO Box 9513, 2300 RA Leiden, The Netherlands bDepartment of Earth Sciences, Utrecht University, Budapestlaan 4, 3584 CD Utrecht, The Netherlands cSchool of Chemistry, The University of Nottingham, University Park, Nottingham NG7 2RD, United Kingdom dDepartment of Physics, Norwegian University of Science and Technology, N-7491 Trondheim, Norway Abstract Triphenylene (C18H12) is a highly symmetric polycyclic aromatic hydrocarbon (PAH) molecule with a `fully-benzenoid' electronic structure. This confers a high chemical stability compared with PAHs of similar size. Although numerous infrared and UV-visible experimental spectroscopic and theoretical studies of a wide range PAHs in an astrophysical context have been conducted, triphenylene and its radical cation have received almost no attention. There exists a huge body of spectroscopic evidence for neutral and ionised PAHs in astrophysical sources, obtained principally through detection of infrared emission features that are characteristic of PAHs as a chemical class. However, it has so far not proved possible to identify spectroscopically a single isolated PAH in space, although PAHs including triphenylene have been detected