Molecular Phylogenetics of the Jay Genus Cyanolyca
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Orden Passeriformes, Familias Malaconotidae a Passeridae)
Ardeol a 57(1), 2010, 199-205 Artículos especiales NOMBRES EN CASTELLANO DE LAS AVES DEL MUNDO RECOMENDADOS POR LA SOCIEDAD ESPAÑOLA DE ORNITOLOGÍA (DECIMOCUARTA PARTE: ORDEN PASSERIFORMES, FAMILIAS MALACONOTIDAE A PASSERIDAE) Eduardo DE JUANA *, Josep DEL HOYO , Manuel FERNÁNDEZ -C RUZ , Xavier FERRER , Ramón SÁEZ -R OYUELA y Jordi SARGATAL INTRODUCCIÓN 110, 51(2): 491-499, 52(2): 389-398, 54(1): 145-153, 56(1): 127-134 y 56(1): 135-142. Esta es la decimocuarta entrega de los nom - En relación a las especies de la avifauna es - bres para las aves del mundo en castellano que pañola, cuando los nombres no coinciden con propone la Sociedad Española de Ornitología los de la Lista Patrón de la Sociedad Españo - (SEO/BirdLife). Tiene su base en el volumen la de Ornitología ( Ardeola , 1: 11-85), en un 14 del Handbook of the Birds of the World apartado final se explican de forma sucinta (Lynx Edicions, Barcelona) , con las siguien - las razones que provocan tales cambios. Son tes 17 familias del orden Passeriformes: Ma - seis especies de córvidos: Garrulus glanda - laconotidae, Prionopidae, Vangidae, Dicruri - rius , Cyanopica cooki , Pica pica , Nucifraga dae, Callaeidae, Notiomystidae, Grallinidae, caryocatactes , Corvus monedul a y Corvus Struthideidae , Artamidae, Cracticidae, Pity - corax . riaseidae, Ptilonorhynchidae, Paradiseidae, Corvidae, Buphagidae, Sturnidae y Passeri - dae. Estas familias agrupan un total de 468 LISTA SISTEMÁTICA DE LOS NOMBRES PROPUESTOS , especies . Como es habitual, la lista sigue en POR FAMILIAS todos sus detalles la clasificación y nomen - clatura científica del mencionado Handbook 176. MALACONOTIDAE (48 especies) of the Birds of the World . -

Printable PDF Format
Field Guides Tour Report Taiwan 2020 Feb 1, 2020 to Feb 12, 2020 Phil Gregory & local guide Arco Huang For our tour description, itinerary, past triplists, dates, fees, and more, please VISIT OUR TOUR PAGE. This gorgeous male Swinhoe's Pheasant was one of the birds of the trip! We found a pair of these lovely endemic pheasants at Dasyueshan. Photo by guide Phil Gregory. This was a first run for the newly reactivated Taiwan tour (which we last ran in 2006), with a new local organizer who proved very good and enthusiastic, and knew the best local sites to visit. The weather was remarkably kind to us and we had no significant daytime rain, somewhat to my surprise, whilst temperatures were pretty reasonable even in the mountains- though it was cold at night at Dasyueshan where the unheated hotel was a bit of a shock, but in a great birding spot, so overall it was bearable. Fog on the heights of Hohuanshan was a shame but at least the mid and lower levels stayed clear. Otherwise the lowland sites were all good despite it being very windy at Hengchun in the far south. Arco and I decided to use a varied assortment of local eating places with primarily local menus, and much to my amazement I found myself enjoying noodle dishes. The food was a highlight in fact, as it was varied, often delicious and best of all served quickly whilst being both hot and fresh. A nice adjunct to the trip, and avoided losing lots of time with elaborate meals. -

Assessing the Habitat Suitability of Six Coniferous Forests Using Avian Assemblages at High Altitude Ecosystem
Assessing The Habitat Suitability of Six Coniferous Forests Using Avian Assemblages At High Altitude Ecosystem M. N. Rajpar Department of Forestry, Shaheed Benazir Bhutto University Sheringal Dir (Upper), Khyber Pakhtunkhwa, Pakistan S. A. Khan Department of Forestry, Shaheed Benazir Bhutto University Sheringal Dir (Upper), Khyber Pakhtunkhwa, Pakistan S. Ullah Department of Forestry, Shaheed Benazir Bhutto University Sheringal Dir (Upper), Khyber Pakhtunkhwa, Pakistan A.H. Rajpar Department of Mechanical Engineering, college of Engineering, Jouf University, Saudi Arabia M. Zakaria ( [email protected] ) Department of Forest Science and Biodiversity, Faculty of Forestry and Environment, Universiti Putra Malaysia Research Article Keywords: birds, conifer forest, density, diversity, habitat, subtropical, vegetation Posted Date: June 15th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-586649/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/20 Abstract Birds are habitat specialist, i.e. often selects habitat that offers ample services such as a variety of food resources, safe breeding and less disturb sites to satisfy their needs. We aimed to ascertain the conservation status of avian assemblages inhabited in six different coniferous forests. The study employed the point count method to determine the avian assemblages in each forest type. The surveys were conducted employing the distance sampling point count method and determining the oristic composition through direct visual estimation from January 2018 to March 2019. In total, 2465 bird individuals representing 57 species and 29 families were observed in six coniferous forests. Twelve tree species, nine shrub species, and 14 grass species were identied in the forests. -

The Relationships of the Starlings (Sturnidae: Sturnini) and the Mockingbirds (Sturnidae: Mimini)
THE RELATIONSHIPS OF THE STARLINGS (STURNIDAE: STURNINI) AND THE MOCKINGBIRDS (STURNIDAE: MIMINI) CHARLESG. SIBLEYAND JON E. AHLQUIST Departmentof Biologyand PeabodyMuseum of Natural History,Yale University, New Haven, Connecticut 06511 USA ABSTRACT.--OldWorld starlingshave been thought to be related to crowsand their allies, to weaverbirds, or to New World troupials. New World mockingbirdsand thrashershave usually been placed near the thrushesand/or wrens. DNA-DNA hybridization data indi- cated that starlingsand mockingbirdsare more closelyrelated to each other than either is to any other living taxon. Some avian systematistsdoubted this conclusion.Therefore, a more extensiveDNA hybridizationstudy was conducted,and a successfulsearch was made for other evidence of the relationshipbetween starlingsand mockingbirds.The resultssup- port our original conclusionthat the two groupsdiverged from a commonancestor in the late Oligoceneor early Miocene, about 23-28 million yearsago, and that their relationship may be expressedin our passerineclassification, based on DNA comparisons,by placing them as sistertribes in the Family Sturnidae,Superfamily Turdoidea, Parvorder Muscicapae, Suborder Passeres.Their next nearest relatives are the members of the Turdidae, including the typical thrushes,erithacine chats,and muscicapineflycatchers. Received 15 March 1983, acceptedI November1983. STARLINGS are confined to the Old World, dine thrushesinclude Turdus,Catharus, Hylocich- mockingbirdsand thrashersto the New World. la, Zootheraand Myadestes.d) Cinclusis -

Costa Rica: the Introtour | July 2017
Tropical Birding Trip Report Costa Rica: The Introtour | July 2017 A Tropical Birding SET DEPARTURE tour Costa Rica: The Introtour July 15 – 25, 2017 Tour Leader: Scott Olmstead INTRODUCTION This year’s July departure of the Costa Rica Introtour had great luck with many of the most spectacular, emblematic birds of Central America like Resplendent Quetzal (photo right), Three-wattled Bellbird, Great Green and Scarlet Macaws, and Keel-billed Toucan, as well as some excellent rarities like Black Hawk- Eagle, Ochraceous Pewee and Azure-hooded Jay. We enjoyed great weather for birding, with almost no morning rain throughout the trip, and just a few delightful afternoon and evening showers. Comfortable accommodations, iconic landscapes, abundant, delicious meals, and our charismatic driver Luís enhanced our time in the field. Our group, made up of a mix of first- timers to the tropics and more seasoned tropical birders, got along wonderfully, with some spying their first-ever toucans, motmots, puffbirds, etc. on this trip, and others ticking off regional endemics and hard-to-get species. We were fortunate to have several high-quality mammal sightings, including three monkey species, Derby’s Wooly Opossum, Northern Tamandua, and Tayra. Then there were many www.tropicalbirding.com +1-409-515-9110 [email protected] Page Tropical Birding Trip Report Costa Rica: The Introtour | July 2017 superb reptiles and amphibians, among them Emerald Basilisk, Helmeted Iguana, Green-and- black and Strawberry Poison Frogs, and Red-eyed Leaf Frog. And on a daily basis we saw many other fantastic and odd tropical treasures like glorious Blue Morpho butterflies, enormous tree ferns, and giant stick insects! TOP FIVE BIRDS OF THE TOUR (as voted by the group) 1. -

Reconstructing the Geographic Origin of the New World Jays
Neotropical Biodiversity ISSN: (Print) 2376-6808 (Online) Journal homepage: http://www.tandfonline.com/loi/tneo20 Reconstructing the geographic origin of the New World jays Sumudu W. Fernando, A. Townsend Peterson & Shou-Hsien Li To cite this article: Sumudu W. Fernando, A. Townsend Peterson & Shou-Hsien Li (2017) Reconstructing the geographic origin of the New World jays, Neotropical Biodiversity, 3:1, 80-92, DOI: 10.1080/23766808.2017.1296751 To link to this article: https://doi.org/10.1080/23766808.2017.1296751 © 2017 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group Published online: 05 Mar 2017. Submit your article to this journal Article views: 956 View Crossmark data Citing articles: 2 View citing articles Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=tneo20 Neotropical Biodiversity, 2017 Vol. 3, No. 1, 80–92, https://doi.org/10.1080/23766808.2017.1296751 Reconstructing the geographic origin of the New World jays Sumudu W. Fernandoa* , A. Townsend Petersona and Shou-Hsien Lib aBiodiversity Institute and Department of Ecology and Evolutionary Biology, University of Kansas, Lawrence, KS, USA; bDepartment of Life Science, National Taiwan Normal University, Taipei, Taiwan (Received 23 August 2016; accepted 15 February 2017) We conducted a biogeographic analysis based on a dense phylogenetic hypothesis for the early branches of corvids, to assess geographic origin of the New World jay (NWJ) clade. We produced a multilocus phylogeny from sequences of three nuclear introns and three mitochondrial genes and included at least one species from each NWJ genus and 29 species representing the rest of the five corvid subfamilies in the analysis. -

India: Kaziranga National Park Extension
INDIA: KAZIRANGA NATIONAL PARK EXTENSION FEBRUARY 22–27, 2019 The true star of this extension was the Indian One-horned Rhinoceros (Photo M. Valkenburg) LEADER: MACHIEL VALKENBURG LIST COMPILED BY: MACHIEL VALKENBURG VICTOR EMANUEL NATURE TOURS, INC. 2525 WALLINGWOOD DRIVE, SUITE 1003 AUSTIN, TEXAS 78746 WWW.VENTBIRD.COM INDIA: KAZIRANGA NATIONAL PARK EXTENSION February 22–27, 2019 By Machiel Valkenburg This wonderful Kaziranga extension was part of our amazing Maharajas’ Express train trip, starting in Mumbai and finishing in Delhi. We flew from Delhi to Guwahati, located in the far northeast of India. A long drive later through the hectic traffic of this enjoyable country, we arrived at our lodge in the evening. (Photo by tour participant Robert Warren) We enjoyed three full days of the wildlife and avifauna spectacles of the famous Kaziranga National Park. This park is one of the last easily accessible places to find the endangered Indian One-horned Rhinoceros together with a healthy population of Asian Elephant and Asiatic Wild Buffalo. We saw plenty individuals of all species; the rhino especially made an impression on all of us. It is such an impressive piece of evolution, a serious armored “tank”! On two mornings we loved the elephant rides provided by the park; on the back of these attractive animals we came very close to the rhinos. The fertile flood plains of the park consist of alluvial silts, exposed sandbars, and riverine flood-formed lakes called Beels. This open habitat is not only good for mammals but definitely a true gem for some great birds. Interesting but common birds included Bar-headed Goose, Red Junglefowl, Woolly-necked Stork, and Lesser Adjutant, while the endangered Greater Adjutant and Black-necked Stork were good hits in the stork section. -

Zoologische Verhandelingen
Systematic notes on Asian birds. 45. Types of the Corvidae E.C. Dickinson, R.W.R.J. Dekker, S. Eck & S. Somadikarta With contributions by M. Kalyakin, V. Loskot, H. Morioka, C. Violani, C. Voisin & J-F. Voisin Dickinson, E.C., R.W.R.J. Dekker, S. Eck & S. Somadikarta. Systematic notes on Asian birds. 45. Types of the Corvidae. Zool. Verh. Leiden 350, 26.xi.2004: 111-148.— ISSN 0024-1652/ISBN 90-73239-95-8. Edward C. Dickinson, c/o The Trust for Oriental Ornithology, Flat 3, Bolsover Court, 19 Bolsover Road, Eastbourne, East Sussex, BN20 7JG, U.K. (e-mail: [email protected]). René W.R.J. Dekker, National Museum of Natural History, P.O. Box 9517, 2300 RA Leiden, The Netherlands (e-mail: [email protected]). Siegfried Eck, Staatliche Naturhistorische Sammlungen Dresden, Museum für Tierkunde, A.B. Meyer Bau, Königsbrücker Landstrasse 159, D-01109 Dresden, Germany (e-mail: [email protected]. sachsen.de). Soekarja Somadikarta, Dept. of Biology, Faculty of Science and Mathematics, University of Indonesia, Depok Campus, Depok 16424, Indonesia (e-mail: [email protected]). Mikhail V. Kalyakin, Zoological Museum, Moscow State University, Bol’shaya Nikitskaya Str. 6, Moscow, 103009, Russia (e-mail: [email protected]). Vladimir M. Loskot, Department of Ornithology, Zoological Institute, Russian Academy of Science, St. Petersburg, 199034 Russia (e-mail: [email protected]). Hiroyuki Morioka, Curator Emeritus, National Science Museum, Hyakunin-cho 3-23-1, Shinjuku-ku, Tokyo 100, Japan. Carlo Violani, Department of Biology, University of Pavia, Piazza Botta 9, 27100 Pavia, Italy (e-mail: [email protected]). -

Appendix, French Names, Supplement
685 APPENDIX Part 1. Speciesreported from the A.O.U. Check-list area with insufficient evidencefor placementon the main list. Specieson this list havebeen reported (published) as occurring in the geographicarea coveredby this Check-list.However, their occurrenceis considered hypotheticalfor one of more of the following reasons: 1. Physicalevidence for their presence(e.g., specimen,photograph, video-tape, audio- recording)is lacking,of disputedorigin, or unknown.See the Prefacefor furtherdiscussion. 2. The naturaloccurrence (unrestrained by humans)of the speciesis disputed. 3. An introducedpopulation has failed to becomeestablished. 4. Inclusionin previouseditions of the Check-listwas basedexclusively on recordsfrom Greenland, which is now outside the A.O.U. Check-list area. Phoebastria irrorata (Salvin). Waved Albatross. Diornedeairrorata Salvin, 1883, Proc. Zool. Soc. London, p. 430. (Callao Bay, Peru.) This speciesbreeds on Hood Island in the Galapagosand on Isla de la Plata off Ecuador, and rangesat seaalong the coastsof Ecuadorand Peru. A specimenwas takenjust outside the North American area at Octavia Rocks, Colombia, near the Panama-Colombiaboundary (8 March 1941, R. C. Murphy). There are sight reportsfrom Panama,west of Pitias Bay, Dari6n, 26 February1941 (Ridgely 1976), and southwestof the Pearl Islands,27 September 1964. Also known as GalapagosAlbatross. ThalassarchechrysosWma (Forster). Gray-headed Albatross. Diornedeachrysostorna J. R. Forster,1785, M6m. Math. Phys. Acad. Sci. Paris 10: 571, pl. 14. (voisinagedu cerclepolaire antarctique & dansl'Ocean Pacifique= Isla de los Estados[= StatenIsland], off Tierra del Fuego.) This speciesbreeds on islandsoff CapeHorn, in the SouthAtlantic, in the southernIndian Ocean,and off New Zealand.Reports from Oregon(mouth of the ColumbiaRiver), California (coastnear Golden Gate), and Panama(Bay of Chiriqu0 are unsatisfactory(see A.O.U. -

The Puzzling Vocal Repertoire of the South American Collared Jay, Cyanolyca Iridicyana Merida
THE PUZZLING VOCAL REPERTOIRE OF THE SOUTH AMERICAN COLLARED JAY, CYANOLYCA VZRIDZCYANA MERIDA JOHN WILLIAM HARDY Previously, I have indicated (Kansas Sci. Bull., 42: 113, 1961) that the New World jays could be systematically grouped in two tribes on the basis of a variety of morphological and behavioral characteristics. I subsequently demonstrated (Occas. Papers Adams Ctr. Ecol. Studies, No. 11, 1964) the relationships between Cyanolyca and Aphelocoma. The tribe Aphelocomini, comprising the currently recognized genera Aphelocoma and Cyanolyca, may be characterized as follows: pattern and ornamen- tation of head simple, consisting of a dark mask, pale superciliary lineation, and no tendency for a crest; tail plain-tipped; vocal repertoire of one to three basic com- ponents, including alarm calls, flock-social calls, or both, that are nasal, querulous, and upwardly or doubly inflected. In contrast, the Cyanocoracini may be charac- tized as follows: pattern of head complex, consisting of triangular cheek patch, super- ciliary spot, and tendency for a crest; tail usually pale-tipped; vocal repertoire usu- ally complex, commonly including a downwardly inflected cuwing call and never an upwardly inflected, nasal querulous call (when repertoire is limited it almost always includes the cazo!ingcall). None of the four Mexican aphelocomine jays has more than three basic calls in its vocal repertoire. There are two exclusively South American species of CyanuZyca (C. viridicyana and C. pulchra). On the basis of the consistency with which the Mexican and Central American speciesof this genus hew to a general pattern of vocalizations as I described it for the tribe, I inferred (1964 op. -
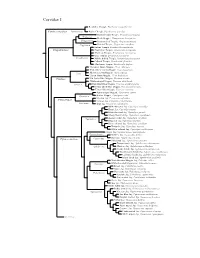
Corvidae Species Tree
Corvidae I Red-billed Chough, Pyrrhocorax pyrrhocorax Pyrrhocoracinae =Pyrrhocorax Alpine Chough, Pyrrhocorax graculus Ratchet-tailed Treepie, Temnurus temnurus Temnurus Black Magpie, Platysmurus leucopterus Platysmurus Racket-tailed Treepie, Crypsirina temia Crypsirina Hooded Treepie, Crypsirina cucullata Rufous Treepie, Dendrocitta vagabunda Crypsirininae ?Sumatran Treepie, Dendrocitta occipitalis ?Bornean Treepie, Dendrocitta cinerascens Gray Treepie, Dendrocitta formosae Dendrocitta ?White-bellied Treepie, Dendrocitta leucogastra Collared Treepie, Dendrocitta frontalis ?Andaman Treepie, Dendrocitta bayleii ?Common Green-Magpie, Cissa chinensis ?Indochinese Green-Magpie, Cissa hypoleuca Cissa ?Bornean Green-Magpie, Cissa jefferyi ?Javan Green-Magpie, Cissa thalassina Cissinae ?Sri Lanka Blue-Magpie, Urocissa ornata ?White-winged Magpie, Urocissa whiteheadi Urocissa Red-billed Blue-Magpie, Urocissa erythroryncha Yellow-billed Blue-Magpie, Urocissa flavirostris Taiwan Blue-Magpie, Urocissa caerulea Azure-winged Magpie, Cyanopica cyanus Cyanopica Iberian Magpie, Cyanopica cooki Siberian Jay, Perisoreus infaustus Perisoreinae Sichuan Jay, Perisoreus internigrans Perisoreus Gray Jay, Perisoreus canadensis White-throated Jay, Cyanolyca mirabilis Dwarf Jay, Cyanolyca nanus Black-throated Jay, Cyanolyca pumilo Silvery-throated Jay, Cyanolyca argentigula Cyanolyca Azure-hooded Jay, Cyanolyca cucullata Beautiful Jay, Cyanolyca pulchra Black-collared Jay, Cyanolyca armillata Turquoise Jay, Cyanolyca turcosa White-collared Jay, Cyanolyca viridicyanus -

Two Nests of the Azure-Hooded Jay with Notes on Nest Attendance
134 THE WILSON BULLETIN l Vol. ZOO,No. I, March 1988 On day 9, we observed a red squirrel (Tumiasciurushudsonicus) prey upon the nestlings in nest B. In most Black-throated Blue Warbler nests, nestlings fledge prematurely ifdisturbed after day 7. The fact that the nestlings could not escape the squirrel’s attack indicates their retarded state of growth. A second clutch (nest C) was initiated by Fl and the polygynous male on 29 June. Four eggs in this third nest hatched on 14 July and feeding duties appeared to be shared equally by the male and female. The four young in nest C fledged on 24 July. Thus, this male was presumably responsible for fertilizing 13 eggs, 12 of which hatched, and seven of which resulted in fledged young. Of 20 Black-throated Blue Warbler pairs closely monitored at Hubbard Brook in 1986, this was the only case of polygyny we found. Monogamy is thought to occur in birds when parental care is needed to successfully raise the young (Lack, Ecological Adaptations for Breeding in Birds, Methuen, New York, New York, 1968). Our observations of the failure of nest B support this contention. Additional work is required to explain the proximate causes of this reproductive strategy. Acknowledgments.-This study was supported by a grant from the National Science Foun- dation to Dartmouth College. We acknowledge the Northeast Forest Experimental Station, U.S. Forest Service for allowing us to work at Hubbard Brook, and K. L. Bildstein, P. A. Gowaty, D. R. Petit, L. J. Petit, N. L. Rodenhouse, T.