Crystal Structure of Dopamine D1 Receptor in Complex with G Protein and a Non-Catechol Agonist
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Product List March 2019 - Page 1 of 53
Wessex has been sourcing and supplying active substances to medicine manufacturers since its incorporation in 1994. We supply from known, trusted partners working to full cGMP and with full regulatory support. Please contact us for details of the following products. Product CAS No. ( R)-2-Methyl-CBS-oxazaborolidine 112022-83-0 (-) (1R) Menthyl Chloroformate 14602-86-9 (+)-Sotalol Hydrochloride 959-24-0 (2R)-2-[(4-Ethyl-2, 3-dioxopiperazinyl) carbonylamino]-2-phenylacetic 63422-71-9 acid (2R)-2-[(4-Ethyl-2-3-dioxopiperazinyl) carbonylamino]-2-(4- 62893-24-7 hydroxyphenyl) acetic acid (r)-(+)-α-Lipoic Acid 1200-22-2 (S)-1-(2-Chloroacetyl) pyrrolidine-2-carbonitrile 207557-35-5 1,1'-Carbonyl diimidazole 530-62-1 1,3-Cyclohexanedione 504-02-9 1-[2-amino-1-(4-methoxyphenyl) ethyl] cyclohexanol acetate 839705-03-2 1-[2-Amino-1-(4-methoxyphenyl) ethyl] cyclohexanol Hydrochloride 130198-05-9 1-[Cyano-(4-methoxyphenyl) methyl] cyclohexanol 93413-76-4 1-Chloroethyl-4-nitrophenyl carbonate 101623-69-2 2-(2-Aminothiazol-4-yl) acetic acid Hydrochloride 66659-20-9 2-(4-Nitrophenyl)ethanamine Hydrochloride 29968-78-3 2,4 Dichlorobenzyl Alcohol (2,4 DCBA) 1777-82-8 2,6-Dichlorophenol 87-65-0 2.6 Diamino Pyridine 136-40-3 2-Aminoheptane Sulfate 6411-75-2 2-Ethylhexanoyl Chloride 760-67-8 2-Ethylhexyl Chloroformate 24468-13-1 2-Isopropyl-4-(N-methylaminomethyl) thiazole Hydrochloride 908591-25-3 4,4,4-Trifluoro-1-(4-methylphenyl)-1,3-butane dione 720-94-5 4,5,6,7-Tetrahydrothieno[3,2,c] pyridine Hydrochloride 28783-41-7 4-Chloro-N-methyl-piperidine 5570-77-4 -

Modulation by Trace Amine-Associated Receptor 1 of Experimental Parkinsonism, L-DOPA Responsivity, and Glutamatergic Neurotransmission
The Journal of Neuroscience, October 14, 2015 • 35(41):14057–14069 • 14057 Neurobiology of Disease Modulation by Trace Amine-Associated Receptor 1 of Experimental Parkinsonism, L-DOPA Responsivity, and Glutamatergic Neurotransmission Alexandra Alvarsson,1* Xiaoqun Zhang,1* Tiberiu L Stan,1 Nicoletta Schintu,1 Banafsheh Kadkhodaei,2 Mark J. Millan,3 Thomas Perlmann,2,4 and Per Svenningsson1 1Department of Clinical Neuroscience, Center for Molecular Medicine, Karolinska Institutet, SE-17176 Stockholm, Sweden, 2Ludwig Institute for Cancer Research, SE-17177 Stockholm, Sweden, 3Pole of Innovation in Neuropsychiatry, Institut de Recherches Servier, Centre de Recherches de Croissy, Paris 87290, France, and 4Department of Cell and Molecular Biology, Karolinska Institutet, SE-17177 Stockholm, Sweden Parkinson’s disease (PD) is a movement disorder characterized by a progressive loss of nigrostriatal dopaminergic neurons. Restoration of dopamine transmission by L-DOPA relieves symptoms of PD but causes dyskinesia. Trace Amine-Associated Receptor 1 (TAAR1) modulates dopaminergic transmission, but its role in experimental Parkinsonism and L-DOPA responses has been neglected. Here, we report that TAAR1 knock-out (KO) mice show a reduced loss of dopaminergic markers in response to intrastriatal 6-OHDA administra- tion compared with wild-type (WT) littermates. In contrast, the TAAR1 agonist RO5166017 aggravated degeneration induced by intra- striatal6-OHDAinWTmice.Subchronic L-DOPAtreatmentofTAAR1KOmiceunilaterallylesionedwith6-OHDAinthemedialforebrain bundle resulted in more pronounced rotational behavior and dyskinesia than in their WT counterparts. The enhanced behavioral sensitization to L-DOPA in TAAR1 KO mice was paralleled by increased phosphorylation of striatal GluA1 subunits of AMPA receptors. Conversely, RO5166017 counteracted both L-DOPA-induced rotation and dyskinesia as well as AMPA receptor phosphorylation. -

The Use of Illicit Drugs As Self-Medication in the Treatment of Cluster Headache: Results from an Italian Online Survey
XML Template (2015) [21.4.2015–2:34pm] [1–5] //blrnas3.glyph.com/cenpro/ApplicationFiles/Journals/SAGE/3B2/CEPJ/Vol00000/150048/APPFile/SG-CEPJ150048.3d (CEP) [PREPRINTER stage] Original Article Cephalalgia 0(0) 1–5 ! International Headache Society 2015 The use of illicit drugs as self-medication Reprints and permissions: sagepub.co.uk/journalsPermissions.nav in the treatment of cluster headache: DOI: 10.1177/0333102415583145 Results from an Italian online survey cep.sagepub.com C Di Lorenzo1, G Coppola2, G Di Lorenzo3, M Bracaglia4, P Rossi5 and F Pierelli4,6 Abstract Background: Cluster headache (CH) patients often receive unsatisfactory treatment and may explore illicit substances as alternatives. We aimed to explore this use of illicit drugs for CH treatment. Methods: We invited CH patients from an Internet-based self-help group to complete a questionnaire regarding their therapeutic use of illicit substances. Results: Of the 54 respondents, 29 were classified as chronic and 39 were drug-resistant cases. Fifty patients had previously tried subcutaneous sumatriptan, 40 had tried O2, and 48 had tried at least one prophylactic treatment. All 54 patients specified that they were dissatisfied with conventional treatments. Thirty-four patients had used cannabin- oids, 13 cocaine, 8 heroin, 18 psilocybin, 12 lysergic acid amide (LSA), and 4 lysergic acid diethylamide (LSD). Discussion: Some patients with intractable CH decided to try illicit drugs concomitantly with cessation of medical care. Most of these patients found suggestions for illicit drug use on the Internet. Many patients seemed to underestimate the judicial consequences of, and had an overestimated confidence in the safety of, such illicit treatments. -

Endogenous Metabolites in Drug Discovery: from Plants to Humans
Endogenous Metabolites in Drug Discovery: from Plants to Humans Joaquim Olivés Farrés TESI DOCTORAL UPF / ANY 201 6 DIRECTOR DE LA TESI: Dr. Jordi Mestres CEXS Department The research in this T hesis has been carried out at the Systems Pharmacolo gy Group , within the Research Programme on Biomedical Informatics (GRIB) at the Parc de Recerca Biomèdica de Barcelona (PRBB). The research presented in this T hesis has been supported by Ministerio de Ciencia e Innovación project BIO2014 - 54404 - R and BIO2011 - 26669 . Printing funded by the Fundació IMIM’s program “Convocatòria d'ajuts 2016 per a la finalització de tesis doctorals de la Fundació IMIM.” Agraïments Voldria donar les gràcies a tanta gent que em fa por deixar - me ningú. Però per c omençar haig agrair en especial al meu director la tesi, Jordi Mestres, per donar - me la oportunitat de formar part del seu laboratori i poder desenvolupar aquí el treball que aquí es presenta. A més d’oferir l’ajuda necessària sempre que ha calgut. També haig de donar les gràcies a tots els companys del grup de Farmacologia de Sistemes que he anat coneguent durants tots aquests anys en què he estat aquí, en especial en Xavi, a qui li he preguntat mil coses, en Nikita, pels sdfs que m’ha anat llençant a CTL ink, i la Irene i la Cristina, que els seus treballs també m’ajuden a completar la tesis. I cal agrair també a la resta de companys del laboratori, l’Albert, la Viktoria, la Mari Carmen, l’Andreas, en George, l’Eric i l’Andreu; de Chemotargets, en Ricard i en David; i altres membres del GRIB, com són l’Alfons, en Miguel, en Pau, l’Oriol i la Carina. -

Drug Screen Req. 06.2016.Docx Rev 2 Date 06-16 Page 1 of 2
TRUESDAIL LABORATORIES, INC. EXCELLENCE IN INDEPENDENT TESTING Established 1931 3337 MICHELSON DRIVE, SUITE CN 750 IRVINE, CALIFORNIA 92612 (714) 730-6239 FAX (714) 730-6462 www. truesdail.com DRUG SCREEN REQUEST FORM SAMPLE TYPE: Urine Blood Plasma Serum Collection Date: ______________ Doctor: ______________________________ Animal Name, HIP # or Other I.D.:______________________________________ Species: _________ Sex: ________ Owner’s name:_______________________ Pre-Purchase Screening (performed by LC/MS) LEVEL Ι: Includes only nonsteroidal anti-inflammatory drugs (phenylbutazone, $100.00 oxyphenbutazone, flunixin, naproxen, ketoprofen, firocoxib, diclofenac and meclofenamic acid). (Requires a minimum of 2 mL serum or plasma or 10mL of urine) LEVEL ΙΙ: Includes LEVEL Ι drugs plus testing for Domosedan (detomidine), $175.00 fluphenazine, acepromazine, promazine, chlorpromazine, triflupromazine, imipramine, propionylpromazine, clomipramine, and reserpine. (Requires a minimum of 4 mL of serum or plasma or 15mL of urine) LEVEL ΙΙΙ: Includes LEVEL Ι and ΙΙ drugs plus testing for butorphanol, triamcinolone $250.00 acetonide, betamethasone, dexamethasone, flumethasone, isoflupredone, predisone, methylprednisolone, prednisolone, albuterol, clenbuterol, terbutaline, and pirbuterol. (Requires a minimum of 5 mL of serum or plasma or 20mL of urine) LEVEL ΙV: TOBA Protocol Testing (urine and blood is required for this test) $300.00 (Requires a minimum of 6 mL of serum or plasma and 25mL of urine) *Rush testing is available for Level I, II or III -

Detomidine and Butorphanol for Standing Sedation in a Range of Zoo-Kept Ungulate Species
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Ghent University Academic Bibliography Journal of Zoo and Wildlife Medicine 48(3): 616–626, 2017 Copyright 2017 by American Association of Zoo Veterinarians DETOMIDINE AND BUTORPHANOL FOR STANDING SEDATION IN A RANGE OF ZOO-KEPT UNGULATE SPECIES Tim Bouts, D.V.M., M.Sc., Dip. E.C.Z.M., Joanne Dodds, V.N., Karla Berry, V.N., Abdi Arif, M.V.Sc., Polly Taylor, Vet. M. B., Ph. D., Dip. E.C.V.A.A., Andrew Routh, B. V. Sc., Cert. Zoo. Med., and Frank Gasthuys, D.V.M., Ph. D., Dip. E.C.V.A.A. Abstract: General anesthesia poses risks for larger zoo species, like cardiorespiratory depression, myopathy, and hyperthermia. In ruminants, ruminal bloat and regurgitation of rumen contents with potential aspiration pneumonia are added risks. Thus, the use of sedation to perform minor procedures is justified in zoo animals. A combination of detomidine and butorphanol has been routinely used in domestic animals. This drug combination, administered by remote intramuscular injection, can also be applied for standing sedation in a range of zoo animals, allowing a number of minor procedures. The combination was successfully administered in five species of nondomesticated equids (Przewalski horse [Equus ferus przewalskii; n ¼ 1], onager [Equus hemionus onager; n ¼ 4], kiang [Equus kiang; n ¼ 3], Grevy’s zebra [Equus grevyi; n ¼ 4], and Somali wild ass [Equus africanus somaliensis; n ¼ 7]), with a mean dose range of 0.10–0.17 mg/kg detomidine and 0.07–0.13 mg/kg butorphanol; the white (Ceratotherium simum simum; n ¼ 12) and greater one-horned rhinoceros (Rhinoceros unicornis; n ¼ 4), with a mean dose of 0.015 mg/kg of both detomidine and butorphanol; and Asiatic elephant bulls (Elephas maximus; n ¼ 2), with a mean dose of 0.018 mg/kg of both detomidine and butorphanol. -

PRACTITIONER's GUIDE Perfect for Routine Procedures on Horses That DON't Want to Have Anything to DO with Routine Procedure
DORMOSEDAN GEL is a registered trademark of Orion Corporation and is distributed by Pfizer Inc. ©2010 Pfizer Inc. All rights reserved. DOR0710058 iMpORtANt SAfEty iNfORMAtiON DORMOSEDAN GEL® is contraindicated in horses with known hypersensitivity to detomidine. Intravenous potentiated sulfonamides should not be used in anesthetized or sedated horses, as potentially fatal dysrhythmias may occur. Do not use DORMOSEDAN GEL in horses with pre-existing atrio-ventricular (AV) or sino-atrial (SA) blocks, cardiovascular disease, respiratory disorders, liver or kidney diseases, or in conditions of shock, severe debilitation or stress due to extreme heat, cold, fatigue or high altitude. Appropriate precautions should be taken while handling and using gel dosing syringes, as DORMOSEDAN GEL can be absorbed following direct exposure to skin, eyes or mouth, and may cause irritation. The use of impermeable gloves is advised. Please see the full prescribing information, or go to www.DormosedanGel.com. www.DormosedanGel.com PerfEct for routine procedures on horses hERE’S hOw DORMOSEDAN GEL wORkS: DORMOSEDAN GEL cAN bE ADMiNiStERED by yOuR thAt don’t want tO hAvE anything tO do cLiENtS fOR A wiDE vARiEty Of routiNE procEDuRES. Detomidine is a potent and relatively selective α2-adrenoceptor agonist with a with routine procedures. central effect inhibiting the transmission of norepinephrine-mediated nervous Depending on the horse’s specific needs, DORMOSEDAN GEL® is impulses. In the animal, the level of consciousness is lowered. The well-characterized an effective standing sedation for use prior to stressful situations profile of detomidine at the receptor level explains its predictable, dose-dependent and minor, nonpainful husbandry procedures, including: pharmacological effects. -

The Effect of Dexmedetomidine and Clonidine on the Inflammatory Response in Critical Illness: a Systematic Review of Animal and Human Studies Charles A
Flanders et al. Critical Care (2019) 23:402 https://doi.org/10.1186/s13054-019-2690-4 RESEARCH Open Access The effect of dexmedetomidine and clonidine on the inflammatory response in critical illness: a systematic review of animal and human studies Charles A. Flanders1† , Alistair S. Rocke1†, Stuart A. Edwardson2, J. Kenneth Baillie1 and Timothy S. Walsh1,2,3* Abstract Background: The α2 agonists, dexmedetomidine and clonidine, are used as sedative drugs during critical illness. These drugs may have anti-inflammatory effects, which might be relevant to critical illness, but a systematic review of published literature has not been published. We reviewed animal and human studies relevant to critical illness to summarise the evidence for an anti-inflammatory effect from α2 agonists. Methods: We searched PubMed, the Cochrane library, and Medline. Animal and human studies published in English were included. Broad search terms were used: dexmedetomidine or clonidine, sepsis, and inflammation. Reference lists were screened for additional publications. Titles and abstracts were screened independently by two reviewers and full-text articles obtained for potentially eligible studies. Data extraction used a bespoke template given study diversity, and quality assessment was qualitative. Results: Study diversity meant meta-analysis was not feasible so descriptive synthesis was undertaken. We identified 30 animal studies (caecal ligation/puncture (9), lipopolysaccharide (14), acute lung injury (5), and ischaemia-reperfusion syndrome (5)), and 9 human studies. Most animal (26 dexmedetomidine, 4 clonidine) and all human studies used dexmedetomidine. In animal studies, α2 agonists reduced serum and/or tissue TNFα (20 studies), IL-6 (17 studies), IL-1β (7 studies), NFκB (6 studies), TLR4 (6 studies), and a range of other mediators. -

Travelers' Guideline for Prohibited And/Or Controlled Medicines and Drugs in the UAE and Quantities Permitted
This is an alphabetical list of INCB and MOH&P controlled Narcotics / Psychotropics / Controlled (CD) and Semi Controlled (SCD) Drugs used for medical purposes, their Scheduling and level of restrictions to carry with travllers to the UAE, with specific medical reasons and supporting documents . Allowed Quantity & Documents to be kept with SL # GENERIC NAME - (Alphabetical Order) CATEGORY (Control Level) the traveller 1 (+) – LYSERGIDE (LSD, LSD-25) Psychotropic Schedule I Prohibited Quantity for the period of stay or a maximum one 2 2c-B Psychotropic Schedule II month use whichever is less. Medical prescription and attested medical report is required 3 3-methylfentanyl NARCOTIC SCHEDULE – IV Prohibited 4 3-methylthiofentanyl NARCOTIC SCHEDULE – IV Prohibited 5 4 – Methylaminorex Psychotropic Schedule I Prohibited 6 4-MTA Psychotropic Schedule I Prohibited 7 Acetorphine NARCOTIC SCHEDULE – IV Prohibited 8 Acetyl-alpha-methylfentanyl NARCOTIC SCHEDULE – IV Prohibited Quantity for the period of stay or a maximum one 9 Acetyldihydrocodeine Narcotic Schedule II month use whichever is less. Medical prescription and attested medical report is required Quantity for the period of stay or a maximum one 10 Acetylmethadol NARCOTIC SCHEDULE – I month use whichever is less. Medical prescription and attested medical report is required Quantity for the period of stay or a maximum one 11 Agomelatine SCD month use whichever is less and need a Medical prescription. Quantity for the period of stay or a maximum one 12 Alfentanyl NARCOTIC SCHEDULE – I month use whichever is less. Medical prescription and attested medical report is required 1/31 This is an alphabetical list of INCB and MOH&P controlled Narcotics / Psychotropics / Controlled (CD) and Semi Controlled (SCD) Drugs used for medical purposes, their Scheduling and level of restrictions to carry with travllers to the UAE, with specific medical reasons and supporting documents . -

An Improved Validated Ultra High Pressure Liquid Chromatography Method for Separation of Rotigotine Impurities in Rotigotine Transdermal Patch
Available online a t www.derpharmachemica.com Scholars Research Library Der Pharma Chemica, 2015, 7(5):26-34 (http://derpharmachemica.com/archive.html) ISSN 0975-413X CODEN (USA): PCHHAX An improved validated ultra high pressure liquid chromatography method for separation of Rotigotine impurities in Rotigotine Transdermal Patch *Avinash S. Patil 1, Shakil S. Sait 2, Girish Deshpande 1, Prakashkumar Acharya 1 and Veeravenkata Srikanth Kaki 1 1Mylan Laboratories Ltd., Formulation AR&D, Plot No. 34 A & B, ANRICH Industrial Estate Bollaram Jinnaram (Mandal), Medak District, Hyderabad, India 2Dr. Reddy’s Laboratories Ltd., Generics A R &D, Innovation Plaza, Survey Nos. 42,45,46 & 54, Bachupalli, Qutubllapur, RR dist, AP, India _____________________________________________________________________________________________ ABSTRACT A rapid, specific, sensitive ultra high-performance liquid chromatographic (UPLC) method has been developed for determination of Rotigotine impurities and its degradation products in Rotigotine Transdermal Patches. UPLC was performed on a C18 column with “mobile phase A” consist of pH 10.00 buffer solution while “mobile phase B” consist of Acetonitrile. The mobile phase was pumped in a gradient manner at the flow-rate of 0.4 mL min −1 . Ultraviolet detection was performed at 225 nm. Rotigotine impurities and degradation products along with process impurities were chromatographed with a total run time of 30 minutes. Calibration showed that response of impurities was a linear function of concentration over the range LOQ to 200% of the target concentration (r 2 ≥ 0.999) and the method was validated over this range for precision, accuracy, linearity and specificity. For precision study, percentage relative standard deviation of each impurity was <15% (n = 6). -
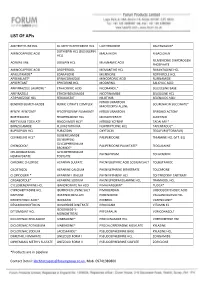
LIST of Apis
LIST OF APIs AMITRIPTYLINE HCL DL-METHYLEPHEDRINE HCL LIOTHYRONINE RALTEGRAVIR* DOTHIEPIN HCL (DOSULEPIN AMINOCAPROIC ACID MALATHION R-BACLOFEN HCL) RILMENIDINE DIHYDROGEN ACRIVASTINE DOXEPIN HCL MEFENAMIC ACID PHOSPHATE AMINOCAPROIC ACID DROPERIDOL MEMANTINE HCL RIMANTADINE HCL APALUTAMIDE* EDARAVONE MILRINONE ROPINIROLE HCL APREMILAST* EFINACONAZOLE MINODRONIC ACID RUFINAMIDE APREPITANT EPHEDRINE HCL MODAFINIL SALICYLIC ACID ARIPIPRAZOLE LAUROXIL* ETHACRYNIC ACID NICORANDIL* SELEGELINE BASE ARIPIRAZOLE ETHOXYBENZAMIDE NICOTINAMIDE SELEGILINE HCL ATIPAMEZOLE HCL FEBUXOSTAT NILOTINIB SELENIUOS ACID NITROFURANTOIN BENDROFLUMETHIAZIDE FERRIC CITRATE COMPLEX SOLIFENACIN SUCCINATE* MACROCRYSTALLINE BENZYL BENZOATE FESOTERODINE FUMARATE NITROFURANTOIN SPIRONOLACTONE BORTEZOMIB FEXOFENADINE HCL MONOHYDRATE SUNITINIB BRETYLIUM TOSYLATE FINGOLIMOD HCL* NITROGLYCERINE TADALAFIL* BRINZOLAMIDE FLUVASTATIN NA NORTRIPTLYINE HCL TAPENTADOL* BUPROPION HCL FURAZIDIN OXYTOCIN TEGAFUR (FTORAFUR) GLIBENCLAMIDE CEVIMELINE HCL* PALIPERIDONE THIAMINE HCL (VIT. B1) (GLYBURIDE) GLYCOPYRRONIUM CHENODIOL* PALIPERIDONE PALMITATE* TIOGUANINE BROMIDE* CHLOROBUTANOL GLYCOPYRRONIUM PHENAZEPAM TOLAZAMIDE HEMIHYDRATE TOSYLATE CHROMIC CHLORIDE HEPARAN SULFATE PHENYLBUTYRIC ACID SODIUM SALT TOLBUTAMIDE CILOSTAZOL HEPARINE CALCIUM PHENYLEPHRINE BITARTRATE TOLCAPONE CLOPIDOGREL* HEPARINE LITHIUM PHENYLEPHRINE HCL TOLTERODINE TARTRATE CRISABOROLE* HEPARINE SODIUM PHENYLPROPANOLAMINE HCL TRAMADOL HCL CYCLOBENZAPRINE HCL IBANDRONATE NA H2O PIMAVANSERIN* TUDCA* CYPROHEPTADINE -

WO 2009/147681 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date 10 December 2009 (10.12.2009) WO 2009/147681 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 45/06 (2006.01) A61K 31/137 (2006.01) kind of national protection available): AE, AG, AL, AM, A61P 25/16 (2006.01) A61K 31/4045 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, A61K 31/135 (2006.01) A61K 31/428 (2006.01) CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, PCT/IL2009/000567 KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, (22) International Filing Date: ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, 7 June 2009 (07.06.2009) NO, NZ, OM, PE, PG, PH, PL, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TJ, TM, TN, TR, TT, (25) Filing Language: English TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/059,326 6 June 2008 (06.06.2008) US GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, (71) Applicant (for all designated States except US): PHAR- TM), European (AT, BE, BG, CH, CY, CZ, DE, DK, EE, MA TWO B LTD.