Naturally Occurring Glycosides and Glycosidases*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Saffron: Chemical Composition and Neuroprotective Activity
molecules Article Saffron: Chemical Composition and Neuroprotective Activity Maria Anna Maggi 1,2,*, Silvia Bisti 3,4 and Cristiana Picco 4,5 1 Hortus Novus srl, via Campo Sportivo 2, 67050 Canistro, Italy 2 Department of Phyisical and Chemical Sciences, University of L’Aquila, Via Vetoio, 67100 Coppito, Italy 3 Department of Biotecnology and Applied Clinical Sciences, DISCAB, University of L’Aquila, Via Vetoio, 67100 Coppito, Italy; [email protected] 4 National Institute of Biostructure and Biosystem (INBB), V. le Medaglie D’Oro 305, 00136 Roma, Italy; [email protected] 5 Institute of Biophysics, National Research Council, Via De Marini 6, 16149 Genova, Italy * Correspondence: [email protected] Academic Editors: Nikolaos Pitsikas and Konstantinos Dimas Received: 14 October 2020; Accepted: 25 November 2020; Published: 29 November 2020 Abstract: Crocus sativus L. belongs to the Iridaceae family and it is commonly known as saffron. The different cultures together with the geoclimatic characteristics of the territory determine a different chemical composition that characterizes the final product. This is why a complete knowledge of this product is fundamental, from which more than 150 chemical compounds have been extracted from, but only about one third of them have been identified. The chemical composition of saffron has been studied in relation to its efficacy in coping with neurodegenerative retinal diseases. Accordingly, experimental results provide evidence of a strict correlation between chemical composition and neuroprotective capacity. We found that saffron’s ability to cope with retinal neurodegeneration is related to: (1) the presence of specific crocins and (2) the contribution of other saffron components. -

Comprehensive Chemotaxonomic Analysis of Saffron Crocus Tepal And
Journal of Pharmaceutical and Biomedical Analysis 184 (2020) 113183 Contents lists available at ScienceDirect Journal of Pharmaceutical and Biomedical Analysis journal homepage: www.elsevier.com/locate/jpba Comprehensive chemotaxonomic analysis of saffron crocus tepal and stamen samples, as raw materials with potential antidepressant activity a b a Javad Mottaghipisheh , Mohammad Mahmoodi Sourestani , Tivadar Kiss , a a c d a,∗ Attila Horváth , Barbara Tóth , Mehdi Ayanmanesh , Amin Khamushi , Dezso˝ Csupor a Department of Pharmacognosy, University of Szeged, Eötvös u. 6, H-6720, Szeged, Hungary b Department of Horticultural Science, Faculty of Agriculture, Shahid Chamran University of Ahvaz, Ahvaz, 61357-43311, Iran c Department of Horticultural Science, Islamic Azad University, Estahban Branch No. 69, Niroo Av., Satarkhan Str., 14536-33143, Tehran, Iran d Department of Horticultural Science, Faculty of Agriculture, University of Mashhad, Mashhad, Iran a r t i c l e i n f o a b s t r a c t Article history: Saffron crocus (Crocus sativus L.) has been widely grown in Iran. Its stigma is considered as the most Received 5 December 2019 valuable spice for which several pharmacological activities have been reported in preclinical and clinical Received in revised form 22 January 2020 studies, the antidepressant effect being the most thoroughly studied and confirmed. This plant part Accepted 17 February 2020 contains several characteristic secondary metabolites, including the carotenoids crocetin and crocin, and Available online 18 February 2020 the monoterpenoid glucoside picrocrocin, and safranal. Since only the stigma is utilized industrially, huge amount of saffron crocus by-product remains unused. Recently, the number of papers dealing with the Keywords: chemical and pharmacological analysis of saffron is increasing; however, there are no systematic studies Crocus sativus Saffron on the chemical variability of the major by-products. -

Geographical Classification of Italian Saffron (Crocus Sativus L.) by Multi-Block Treatments of UV-Vis and IR Spectroscopic Data
molecules Article Geographical Classification of Italian Saffron (Crocus sativus L.) by Multi-Block Treatments of UV-Vis and IR Spectroscopic Data Alessandra Biancolillo * , Martina Foschi and Angelo Antonio D’Archivio Department of Physical and Chemical Sciences, University of L’Aquila, Via Vetoio Coppito, 67100 L’Aquila, Italy; [email protected] (M.F.); [email protected] (A.A.D.) * Correspondence: [email protected] Academic Editors: Emanuela Zanardi, Lenka Husáková and Jose Miguel Hernandez-Hierro Received: 27 April 2020; Accepted: 15 May 2020; Published: 16 May 2020 Abstract: One-hundred and fourteen samples of saffron harvested in four different Italian areas (three in Central Italy and one in the South) were investigated by IR and UV-Vis spectroscopies. Two different multi-block strategies, Sequential and Orthogonalized Partial Least Squares Linear Discriminant Analysis (SO-PLS-LDA) and Sequential and Orthogonalized Covariance Selection Linear Discriminant Analysis (SO-CovSel-LDA), were used to simultaneously handle the two data blocks and classify samples according to their geographical origin. Both multi-block approaches provided very satisfying results. Each model was investigated in order to understand which spectral variables contribute the most to the discrimination of samples, i.e., to the characterization of saffron harvested in the four different areas. The most accurate solution was provided by SO-PLS-LDA, which only misclassified three test samples over 31 (in external validation). Keywords: saffron; infrared; ultraviolet; classification; multi-block; data fusion; SO-PLS; SO-CovSel 1. Introduction Saffron, the dried stigma of Crocus sativus L., has been used since ancient times as a spice, food dye, or herbal medicinal. -

Type of the Paper (Article
Nutrients 2019: Nutritional Advances in the Prevention and Management of Chronic Disease AXA Convention Centre Barcelona, Spain 25 – 27 September 2019 Conference Chairs Prof. Lluis Serra Majem Prof. María Luz Fernández Organised by Conference Secretariat Sara Martínez Pablo Velázquez Facundo Santomé Xiaocen Zhang Lucia Russo Nutrients 2019 Conference 1 #Nutrients2019 CONTENTS Abridged Programme 5 Conference Programme 6 Welcome 15 General Information 17 Abstracts – Session 1 27 Nutritional Advances in the Prevention and Management of Chronic Disease Abstracts – Session 2 63 Nutrients and the Microbiome Abstracts – Special Session 73 Cognitive and Mental Health (sponsored by the California Walnuts Commission) Abstracts – Session 3 77 Bioactive/Functional Foods (sponsored by the journal Foods) Abstracts – Session 4 97 Mediterranean Diet Abstracts – Session 5 117 Nutrient and Lifestyle Interactions Abstracts – Session 6 127 Nutrient for Ageing Populations Abstracts – Session 7 135 Polyphenols and Antioxidants Abstracts – Session 8 149 Dietary Assessment Abstracts – Poster Exhibition 163 Nutrients 2019 Conference 3 Nutrients 2019 – Nutritional Advances in the Prevention and Management of Chronic Disease 25 – 27 September 2019, Barcelona, Spain Wednesday Thursday Friday 25 September 2019 26 September 2019 27 September 2019 Session 2 Check-in Session 5 Special Session Opening Ceremony Coffee Break Coffee Break Morning Session 6 Session 1. Part I Session 3 Session 7 Lunch & Lunch & Lunch Poster Session A Poster Session B Session 1. Part II Session -

Crocus Sativus), a Species of Crocus in the Family Iridaceae
1 CHAPTERl INTRODUCTION 1.1 Background of Study Saffron is a spice derived from the flower of the saffron crocus (Crocus sativus), a species of crocus in the family Iridaceae. Saffron, which has for decades been the world's most expensive spice by weight, is native to Southwest Asia. Saffron a spice derived from the dried stigma of the saffron and saffron also known as crocus sativus among the chemist or simply its scientific name is crocus sativus. (Abdulalev et.al.2006) Saffron is native to Southwest Asia but first was cultivated in Greece. In late history of Egypt, they used a quarter cup of saffron to mix with warm water for bath because of its coloring and cosmetic properties.Egyptian used saffron as a treatment for all varieties of gastrointestinal ailments. Saffron was used by ancient Persian worshipers as a ritual offering to deities. It was also used as a brilliant yellow dye, a perfume, and a medicine. Various conflicting accounts exist that describe saffron's first arrival in South and East Asia. The first of these rely on historical accounts gleaned from Persian records. (Abdulalev et.al.2006) Saffron is dry, glossy and greasy to the touch when freshly dried, turning dull and brittle with age. It is easily bleached if not stored in the dark, and also stores better under conditions of low temperature and low relative humidity. (Rangahau Mana Kai.2003).The chemistry of saffron has been investigated in detail. The major pigment, a water-soluble carotenoid giving saffron its value as a dye, is crocin, a yellow-red pigment found at levels of up to 2%. -

Case Studies Exploring the Impact of Synthetic Biology on Natural
Synthetic Biology, Biodiversity & Farmers www.etcgroup.org Case studies exploring the impact of synthetic biology on natural products, livelihoods and sustainable use of biodiversity Synthetic Biology, Biodiversity and Farmers ETC Group ...works to address the socioeconomic and ecological issues surrounding new technologies that could have an impact on the world’s poorest and most vulnerable people. We investigate ecological erosion (including the erosion of cultures and human rights); the development of new technologies (especially agricultural but also other technologies that work with genomics and matter); and we monitor global Synthetic Biology, Biodiversity and Farmers governance issues including corporate concentration ...is ETC Communiqué #116 and trade in technologies. We operate at the global Published April 2016 political level. We work closely with partner civil Editing by Holly Dressel society organizations (CSOs) and social movements, Additional case study research by Marie-Pier Munger, especially in Africa, Asia and Latin America. Kathy Jo Wetter, Hope Shand and Dru Ojay www.etcgroup.org Design and Illustration by Stig ETC is grateful to The Swift Foundation for financial support in the preparation and revision of these case studies. 2 Synthetic Biology, Biodiversity and Farmers Contents 13 Case studies Introduction 3 Agarwood 12 What is Synthetic Biology? 4 Ambergris / Clary sage 16 4 The Switch to Ingredient Markets Artemisinin 19 Metabolic Pathways to Everything 4 Ginseng 22 Industry’s Synbio Advantage 6 Patchouli 25 Answering -

Saffron Flavor: Compounds Involved, Biogenesis and Human Perception
® Functional Plant Science and Biotechnology ©2010 Global Science Books Saffron Flavor: Compounds Involved, Biogenesis and Human Perception Luana Maggi • Manuel Carmona • Ana M. Sánchez • Gonzalo L. Alonso* Cátedra de Química Agrícola E.T.S.I. Agrónomos de Albacete, Universidad de Castilla La Mancha, 02071 Albacete, Spain Corresponding author : * [email protected] ABSTRACT In recent years, saffron has increased in interest for both scientists and consumers, as it is the only spice able to give to food flavor, color and aroma to foods. In relation to flavor, picrocrocin is considered as mainly responsible for saffron’s bitter taste, but other compounds structurally related to picrocrocin and flavonoids have been identified and could contribute to such a property. Further studies are necessary to establish picrocrocin sensory perception, as only its taste detection threshold has been established (10 mgL-1). Even though it is well known that picrocrocin content is directly affected by the dehydration process of the spice, its generation pathway remains unclear. In this paper a comparison between the classical hypothesis and the alternative one is therefore presented. Among its flavor properties, picrocrocin is an excellent marker of saffron purity because till now its presence it is only reported in saffron. Also, it is especially useful in unmasking sophisticated adulterations carried out with pigments from Gardenia jasminoides, which contain the same carotenoid family as saffron. For both reasons, it is important to accurately determine picrocrocin content in order to gain the trust of saffron dealers and also for consumer satisfaction. This review summarizes the available methodologies for this proposal, giving emphasis to the gaps contained in the current ISO 3632 Standard, normally used in the international market. -

Estimation of Alkaloid, Saponin and Flavonoid, Content in Various
Journal of Medicinal Plants Studies 2016; 4(5): 171-174 ISSN 2320-3862 JMPS 2016; 4(5): 171-174 Estimation of alkaloid, saponin and flavonoid, © 2016 JMPS Received: 23-07-2016 content in various extracts of Crocus sativa Accepted: 24-08-2016 M Amin Mir M Amin Mir, Kajal Parihar, Uzma Tabasum and Ekata Kumari Dept. of Chemistry Uttaranchal College of Science and Technology, Dehradun, Abstract Uttarakhand, India Crocus sativa is a well-known spice grown mostly in the Kashmir region of India. The flowers of the plant have a good aroma and are full of secondary metabolites due to which the flowers are being used Kajal Parihar dying agents and the flavoring agents in almost all types of dishes. The flowers of the concerned plant Dept. of Botany have been analysed for the estimation of Alkaloid, Saponin and Flavonoid contents. Three solvents viz Uttaranchal College of Science DCM, Methanol and Water have been used for the extraction of Alkaloid, Saponin and Flavonoid and Technology, Dehradun, contents. Different solvent extracts extract different concentration of metabolites. The alkaloid content Uttarakhand, India was found to be (6.4 and 2.4mg/g) in the methanol and water extract of the flowers, similarly the flavonoid content was found to be 1.8, 9.2 and 11.2mg/g in DCM, Methanol and water extract Uzma Tabasum respectively. The Saponin content was found to be 1.2 and 3.4mg/g in the methanol and water extract of Dept. of Botany the crocus sativa. Uttaranchal College of Science and Technology, Dehradun, Keywords: Alkaloids, saponin, flavonoids, crocus sativa, DCM, methanol and water Uttarakhand, India Introduction Ekata Kumari Dept. -

Bioactivity and Bioavailability of the Major Metabolites of Crocus Sativus L
molecules Review BioactivityReview and Bioavailability of the Major Bioactivity and Bioavailability of the Major Metabolites of Crocus sativus L. Flower Metabolites of Crocus sativus L. Flower Natalia Moratalla-López , María José Bagur, Cándida Lorenzo, M.E. Martínez-Navarro, M.Natalia Rosario Moratalla-López, Salinas and María Gonzalo José L. Bagur, Alonso Cándida * Lorenzo, M.E. Martínez-Navarro, M.Cá Rosariotedra de QuSalinasímica Agrandí cola,Gonzalo ETSI AgrL. Alonsoónomos * y de Montes, Universidad de Castilla-La Mancha, CátedraCampus de Universitario, Química Agrícola, 02071 Albacete,ETSI Agrónomos Spain y de Montes, Universidad de Castilla-La Mancha, Campus Universitario,* Correspondence: 02071 Albacete, [email protected]; Spain Tel.: +34 967 599210; Fax: +34 967 599238 * Correspondence: [email protected]; Tel.: +34 967 599210; Fax: +34 967 599238 Received: 16 July 2019; Accepted: 30 July 2019; Published: 2 August 2019 Received: 16 July 2019; Accepted: 30 July 2019; Published: date Abstract: Crocus sativus L. has been cultivated throughout history to obtain its flowers, whose dried stigmasAbstract: give Crocus rise sativus to the L. spice has knownbeen cultivated as saffron. throughout Crocetin history esters, to picrocrocin, obtain its flowers, and safranal whose are dried the mainstigmas metabolites give rise ofto thisthe spice,spice known which possessas saffron. a great Crocetin bioactivity, esters, although picrocrocin, the mechanisms and safranal of are action the andmain its metabolites bioavailability of this are spice, still towh beich solved. possess The a great rest ofbioactivity, the flower al isthough composed the mechanisms by style, tepals, of action and stamensand its bioavailability that have other are compounds, still to be solved. -
Chemical Composition of Saffron: a Review Syed Muzaffar*A, Tanveer A
Int J Biol Med Res.2019 ;10(4):6910 - 6919 Int J Biol Med Res www.biomedscidirect.com Volume 10, Issue 4, OCT 2019 Contents lists available at BioMedSciDirect Publications International Journal of Biological & Medical Research Journal homepage: www.biomedscidirect.com BioMedSciDirect International Journal of Publications BIOLOGICAL AND MEDICAL RESEARCH Review article Chemical Composition of Saffron: A Review Syed Muzaffar*a, Tanveer A. Sofib and Khaliquz Zaman Khana Department of Chemistry, University of Kashmir, Srinagar 190006, India, 2Department of Zoology, University of Kashmir, Srinagar 190006, India A R T I C L E I N F O A B S T R A C T Keywords: Abstract: Saffron, considered being the king of spices with the costliest tag in terms of weight Saffron has not received much scientific attention within the state of Jammu and Kashmir, though this Jammu and Kashmir Cultivation is an important privileged asset to this state and constitutes approximately 16% of agricultural Chemistry income. This golden crop of our state is world famous due to its glorifying dark reddish aromatic stigmas and there are thousands of people involved in cultivation and trade of saffron within and outside Jammu and Kashmir. More importantly, there is a deep interest growing globally among scientists, herbal and pharmaceutical industries to explore biochemical and biological profile of saffron spice so that various products could be formulated based on saffron integration. Further, there was not much literature available about saffron native to Jammu and Kashmir particularly related to the impact of different drying methods and soil constituents on its chemical profile as well as nutritional values and evaluation of its activities such as anticancer, antibacterial and antifungal. -
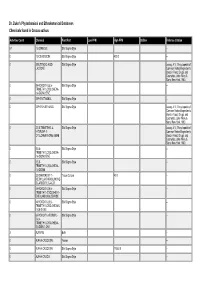
Dr. Duke's Phytochemical and Ethnobotanical Databases Chemicals Found in Crocus Sativus
Dr. Duke's Phytochemical and Ethnobotanical Databases Chemicals found in Crocus sativus Activities Count Chemical Plant Part Low PPM High PPM StdDev Refernce Citation 67 1,8-CINEOLE Silk Stigma Style -- 0 13-CIS-CROCIN Silk Stigma Style 400.0 -- 0 2-BUTENOIC-ACID- Silk Stigma Style Leung, A.Y., Encyclopedia of LACTONE Common Natural Ingredients Used in Food, Drugs, and Cosmetics, John Wiley & Sons, New York, 1980. 0 2-HYDROXY-3,5,5- Silk Stigma Style -- TRIMETHYLCYCLOHEXA- 1,4-DION-2-ENE 0 2-PHENETHANOL Silk Stigma Style -- 0 2-PHENYLETHANOL Silk Stigma Style Leung, A.Y., Encyclopedia of Common Natural Ingredients Used in Food, Drugs, and Cosmetics, John Wiley & Sons, New York, 1980. 0 3,5,5-TRIMETHYL-4- Silk Stigma Style Leung, A.Y., Encyclopedia of HYDROXY-1- Common Natural Ingredients CYCLOHEXANONE-2-ENE Used in Food, Drugs, and Cosmetics, John Wiley & Sons, New York, 1980. 0 3,5,5- Silk Stigma Style -- TRIMETHYLCYCLOHEXA- 1,4-DION-2-ENE 0 3,5,5- Silk Stigma Style -- TRIMETHYLCYCLOHEXA- 1,4-DIONE 0 3,8-DIHYDROXY-1- Tissue Culture 40.0 -- METHYLANTHRAQUINONE- 2-CARBOXYLIC-ACID 0 4-HYDROXY-2,6,6- Silk Stigma Style -- TRIMETHYL-CYCLOHEX-1- ENE-CARBOXALDEHYDE 0 4-HYDROXY-3,5,5- Silk Stigma Style -- TRIMETHYLCYCLOHEXAN- 1-ON-2-ENE 0 6-HYDROXY-3-FORMYL- Silk Stigma Style -- 2,4,4- TRIMETHYLCYCLOHEXA- 2,5-DIEN-1-ONE 3 ALANINE Bulb -- 0 ALPHA-CROCETIN Flower -- 0 ALPHA-CROCETIN Silk Stigma Style 17500.0 -- 0 ALPHA-CROCIN Silk Stigma Style -- Activities Count Chemical Plant Part Low PPM High PPM StdDev Refernce Citation 28 ALPHA-PINENE Silk Stigma Style -- 13 ALPHA-TERPINENE Silk Stigma Style -- 0 ALPHA-THUJENE Silk Stigma Style -- 14 ARGININE Bulb -- 0 ASH Silk Stigma Style 42700.0 133000.0 1.0 ANON. -

Crocus: a Novel Regulator of Apocarotenoid Biosynthesis Nasheeman Ashraf1*, Deepti Jain2 and Ram a Vishwakarma3
Ashraf et al. BMC Plant Biology (2015) 15:25 DOI 10.1186/s12870-015-0423-7 RESEARCH ARTICLE Open Access Identification, cloning and characterization of an ultrapetala transcription factor CsULT1 from Crocus: a novel regulator of apocarotenoid biosynthesis Nasheeman Ashraf1*, Deepti Jain2 and Ram A Vishwakarma3 Abstract Background: Crocus sativus is a triploid sterile plant with long red stigmas which form commercial saffron. Saffron is the site for synthesis and accumulation of apocarotenoids like crocin, picrocrin and safranal which are responsible for its color, flavour and aroma making it world’s most expensive spice. These compounds are formed by oxidative cleavage of zeaxanthin by carotenoid cleavage dioxygenases. Although the biosynthetic pathway of apocarotenoids is known to a considerable extent, the mechanism that regulates its tissue and developmental stage specific expression is not known. Results: In the present work, we identified, cloned and characterized ultrapetala transcription factor called CsULT1 from Crocus. The gene contains an 80 amino acid long conserved SAND domain. The CsULT1 transcript was more abundant in stigma and showed increase in expression from pre anthesis stage till anthesis and decreased in post anthesis stage which corroborated with the accumulation pattern of crocin indicating its possible role in regulation of apocarotenoid biosynthesis. CsULT1 was found to be transcriptionally active and localized in nucleus. Its expression is induced in response to phytohormones like auxin, methyljasmonate and salicylic acid. Overexpression of CsULT1 in Crocus calli resulted in enhanced expression of key pathway genes like phytoene synthase (PSY), phytoene desaturase (PDS), beta carotene hydroxylase (BCH) and carotenoid cleavage dioxygenases (CCDs) indicating its role in regulation of apocarotenoid biosynthesis.