761037Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Old and New Challenges in Uveitis Associated with Behçet's Disease
Journal of Clinical Medicine Review Old and New Challenges in Uveitis Associated with Behçet’s Disease Julie Gueudry 1,* , Mathilde Leclercq 2, David Saadoun 3,4,5 and Bahram Bodaghi 6 1 Department of Ophthalmology, Hôpital Charles Nicolle, F-76000 Rouen, France 2 Department of Internal Medicine, Hôpital Charles Nicolle, F-76000 Rouen, France; [email protected] 3 Department of Internal Medicine and Clinical Immunology, AP-HP, Centre National de Références Maladies Autoimmunes et Systémiques Rares et Maladies Autoinflammatoires Rares, Groupe Hospitalier Pitié-Salpêtrière, F-75013 Paris, France; [email protected] 4 Sorbonne Universités, UPMC Univ Paris 06, INSERM, UMR S 959, Immunology-Immunopathology-Immunotherapy (I3), F-75005 Paris, France 5 Biotherapy (CIC-BTi), Hôpital Pitié-Salpêtrière, AP-HP, F-75651 Paris, France 6 Department of Ophthalmology, IHU FOReSIGHT, Sorbonne-AP-HP, Groupe Hospitalier Pitié-Salpêtrière, F-75013 Paris, France; [email protected] * Correspondence: [email protected]; Tel.: +33-2-32-88-80-57 Abstract: Behçet’s disease (BD) is a systemic vasculitis disease of unknown origin occurring in young people, which can be venous, arterial or both, classically occlusive. Ocular involvement is particularly frequent and severe; vascular occlusion secondary to retinal vasculitis may lead to rapid and severe loss of vision. Biologics have transformed the management of intraocular inflammation. However, the diagnosis of BD is still a major challenge. In the absence of a reliable biological marker, diagnosis is based on clinical diagnostic criteria and may be delayed after the appearance of the onset sign. However, therapeutic management of BD needs to be introduced early in order to control inflammation, to preserve visual function and to limit irreversible structural damage. -

Review Anti-Cytokine Biologic Treatment Beyond Anti-TNF in Behçet's Disease
Review Anti-cytokine biologic treatment beyond anti-TNF in Behçet’s disease A. Arida, P.P. Sfikakis First Department of Propedeutic Internal ABSTRACT and thrombotic complications (1-3). Medicine Laikon Hospital, Athens, Unmet therapeutic needs in Behçet’s Treatment varies according to type and University Medical School, Greece. disease have drawn recent attention to severity of disease manifestations. Cor- Aikaterini Arida, MD biological agents targeting cytokines ticosteroids, interferon-alpha and con- Petros P. Sfikakis, MD other than TNF. The anti-IL-17 anti- ventional immunosuppressive drugs, Please address correspondence to: body secukinumab and the anti-IL-2 such as azathioprine, cyclosporine-A, Petros P. Sfikakis, MD, receptor antibody daclizumab were not cyclophosphamide and methotrexate, First Department of Propedeutic superior to placebo for ocular Behçet’s and Internal Medicine, are used either alone or in combination Laikon Hospital, in randomised controlled trials, com- for vital organ involvement. During the Athens University Medical School, prising 118 and 17 patients, respec- last decade there has been increased use Ag Thoma, 17, tively. The anti-IL-1 agents anakinra of anti-TNF monoclonal antibodies in GR-11527 Athens, Greece. and canakinumab and the anti-IL-6 patients with BD who were refractory E-mail: [email protected] agent tocilizumab were given to iso- to conventional treatment or developed Received on June 7, 2014; accepted in lated refractory disease patients, who life-threatening complications (4, 5). revised form on September 17, 2014. were either anti-TNF naïve (n=9) or Anti-TNF treatment has been shown to Clin Exp Rheumatol 2014; 32 (Suppl. 84): experienced (n=18). -

Ilaris Canakinumab for Systemic Juvenile Idiopathic Arthritis SJIA
Subject: Ilaris (canakinumab) for Systemic Juvenile Original Effective Date: 4/24/2015 Idiopathic Arthritis (SJIA) Policy Number: MCP-246 Revision Date(s): Review Date(s): 12/15/2016; 6/22/2017 DISCLAIMER This Medical Policy is intended to facilitate the Utilization Management process. It expresses Molina's determination as to whether certain services or supplies are medically necessary, experimental, investigational, or cosmetic for purposes of determining appropriateness of payment. The conclusion that a particular service or supply is medically necessary does not constitute a representation or warranty that this service or supply is covered (i.e., will be paid for by Molina) for a particular member. The member's benefit plan determines coverage. Each benefit plan defines which services are covered, which are excluded, and which are subject to dollar caps or other limits. Members and their providers will need to consult the member's benefit plan to determine if there are any exclusion(s) or other benefit limitations applicable to this service or supply. If there is a discrepancy between this policy and a member's plan of benefits, the benefits plan will govern. In addition, coverage may be mandated by applicable legal requirements of a State, the Federal government or CMS for Medicare and Medicaid members. CMS's Coverage Database can be found on the CMS website. The coverage directive(s) and criteria from an existing National Coverage Determination (NCD) or Local Coverage Determination (LCD) will supersede the contents of this Molina medical coverage policy (MCP) document and provide the directive for all Medicare members. SUMMARY This policy addresses the coverage of Ilaris (canakinumab) for the treatment of Systemic Juvenile Idiopathic Arthritis (SJIA) when appropriate criteria are met. -

Inflammatory Conditions – Kevzara™ (Sarilumab for Subcutaneous Injection)
Cigna National Formulary Coverage Policy Prior Authorization Inflammatory Conditions – Kevzara® (sarilumab for subcutaneous injection) Table of Contents Product Identifier(s) National Formulary Medical Necessity ................ 1 59231 Conditions Not Covered....................................... 2 Background .......................................................... 2 References .......................................................... 3 Revision History ................................................... 3 INSTRUCTIONS FOR USE The following Coverage Policy applies to health benefit plans administered by Cigna Companies. Certain Cigna Companies and/or lines of business only provide utilization review services to clients and do not make coverage determinations. References to standard benefit plan language and coverage determinations do not apply to those clients. Coverage Policies are intended to provide guidance in interpreting certain standard benefit plans administered by Cigna Companies. Please note, the terms of a customer’s particular benefit plan document [Group Service Agreement, Evidence of Coverage, Certificate of Coverage, Summary Plan Description (SPD) or similar plan document] may differ significantly from the standard benefit plans upon which these Coverage Policies are based. For example, a customer’s benefit plan document may contain a specific exclusion related to a topic addressed in a Coverage Policy. In the event of a conflict, a customer’s benefit plan document always supersedes the information in the Coverage Policies. In the absence of a controlling federal or state coverage mandate, benefits are ultimately determined by the terms of the applicable benefit plan document. Coverage determinations in each specific instance require consideration of 1) the terms of the applicable benefit plan document in effect on the date of service; 2) any applicable laws/regulations; 3) any relevant collateral source materials including Coverage Policies and; 4) the specific facts of the particular situation. -

Cytokine & CAM Antagonists
Cytokine & CAM Antagonists Medical policy no. 66.27.00-3 Effective Date: October 1, 2019 Note: New-to-market drugs included in this class based on the Apple Health Preferred Drug List are non-preferred and subject to this prior authorization (PA) criteria. Non-preferred agents in this class require an inadequate response or documented intolerance due to severe adverse reaction or contraindication to at least TWO preferred agents. If there is only one preferred agent in the class documentation of inadequate response to ONE preferred agent is needed. If a drug within this policy receives a new indication approved by the Food and Drug Administration (FDA), medical necessity for the new indication will be determined on a case-by-case basis following FDA labeling. To see the list of the current Apple Health Preferred Drug List (AHPDL), please visit: https://www.hca.wa.gov/assets/billers-and- providers/apple-health-preferred-drug-list.xlsx Background: Cytokines and cell-adhesion molecule (CAM) are chemical mediators involved in inflammatory processes throughout the body. Medications included in this policy are used to treat a group of diseases that may be caused or worsened by an overactive immune system such as rheumatoid arthritis, psoriasis, and ulcerative colitis. Administration is different for each medication, and may be administered subcutaneously (SC), intravenously (IV), or orally. Medical necessity Drug Medical Necessity adalimumab (HUMIRA) Cytokine and CAM antagonists may be considered medically necessary etanercept (ENBREL) when -

(12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub
US 20170172932A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0172932 A1 Peyman (43) Pub. Date: Jun. 22, 2017 (54) EARLY CANCER DETECTION AND A 6LX 39/395 (2006.01) ENHANCED IMMUNOTHERAPY A61R 4I/00 (2006.01) (52) U.S. Cl. (71) Applicant: Gholam A. Peyman, Sun City, AZ CPC .......... A61K 9/50 (2013.01); A61K 39/39558 (US) (2013.01); A61K 4I/0052 (2013.01); A61 K 48/00 (2013.01); A61K 35/17 (2013.01); A61 K (72) Inventor: sham A. Peyman, Sun City, AZ 35/15 (2013.01); A61K 2035/124 (2013.01) (21) Appl. No.: 15/143,981 (57) ABSTRACT (22) Filed: May 2, 2016 A method of therapy for a tumor or other pathology by administering a combination of thermotherapy and immu Related U.S. Application Data notherapy optionally combined with gene delivery. The combination therapy beneficially treats the tumor and pre (63) Continuation-in-part of application No. 14/976,321, vents tumor recurrence, either locally or at a different site, by filed on Dec. 21, 2015. boosting the patient’s immune response both at the time or original therapy and/or for later therapy. With respect to Publication Classification gene delivery, the inventive method may be used in cancer (51) Int. Cl. therapy, but is not limited to such use; it will be appreciated A 6LX 9/50 (2006.01) that the inventive method may be used for gene delivery in A6 IK 35/5 (2006.01) general. The controlled and precise application of thermal A6 IK 4.8/00 (2006.01) energy enhances gene transfer to any cell, whether the cell A 6LX 35/7 (2006.01) is a neoplastic cell, a pre-neoplastic cell, or a normal cell. -

Immunomodulators
Therapeutic Class Overview Immunomodulators INTRODUCTION Immunomodulators treat a wide variety of conditions, including rheumatoid arthritis (RA), juvenile idiopathic arthritis (JIA), plaque psoriasis (PsO), psoriatic arthritis (PsA), ankylosing spondylitis (AS), Crohn’s disease (CD), ulcerative colitis (UC), hidradenitis suppurativa (HS), and uveitis (UV), as well as several less common conditions. T cells, B cells, and cytokines such as tumor necrosis factor (TNF), interleukin-1 (IL-1) and interleukin-6 (IL-6) play a key role in the inflammatory and immune process (Choy et al 2001). This has led to the development of biologic agents to target these areas. The Food and Drug Administration (FDA) has currently approved 5 originator TNF inhibitors: Cimzia (certolizumab), Enbrel (etanercept), Humira (adalimumab), Remicade (infliximab), and Simponi/Simponi Aria (golimumab), as well as 7 biosimilar TNF inhibitors: Amjevita (adalimumab-atto), Erelzi (etanercept-szzs), Hyrimoz (adalimumab-adaz), Inflectra (infliximab-dyyb), Renflexis (infliximab-abda), Cyltezo (adalimumab-adbm), and Ixifi (infliximab-qbtx). Other agents targeting different cells and cytokines are also FDA- approved for RA treatment. These include Orencia (abatacept), which inhibits CD28-B7 mediated costimulation of the T-cell; Rituxan (rituximab), which targets CD20, a molecule that is found on the surface of B-cells; Actemra (tocilizumab) and Kevzara (sarilumab), which have activity directed against the IL-6 receptor; and Kineret (anakinra), which targets the IL-1 receptor. Of these agents, one biosimilar product has been approved: Truxima (rituximab- abbs). Oral agents on the market, Xeljanz and Xeljanz XR (tofacitinib) and Olumiant (baricitinib), target Janus- associated kinase (JAK) pathways. By inhibiting the JAK pathway, the ability of cytokines to produce inflammation is reduced. -

Soluble Ligands As Drug Targets
REVIEWS Soluble ligands as drug targets Misty M. Attwood 1, Jörgen Jonsson1, Mathias Rask- Andersen 2 and Helgi B. Schiöth 1,3 ✉ Abstract | Historically, the main classes of drug targets have been receptors, enzymes, ion channels and transporters. However, owing largely to the rise of antibody- based therapies in the past two decades, soluble protein ligands such as inflammatory cytokines have become an increasingly important class of drug targets. In this Review, we analyse drugs targeting ligands that have reached clinical development at some point since 1992. We identify 291 drugs that target 99 unique ligands, and we discuss trends in the characteristics of the ligands, drugs and indications for which they have been tested. In the last 5 years, the number of ligand-targeting drugs approved by the FDA has doubled to 34, while the number of clinically validated ligand targets has doubled to 22. Cytokines and growth factors are the predominant types of targeted ligands (70%), and inflammation and autoimmune disorders, cancer and ophthalmological diseases are the top therapeutic areas for both approved agents and agents in clinical studies, reflecting the central role of cytokine and/or growth factor pathways in such diseases. Drug targets In the twentieth century, drug discovery largely involved far more challenging to achieve with small- molecule Pharmacological targets, such the identification of small molecules that exert their drugs. Protein ligands have been successfully targeted as proteins, that mediate the therapeutic effects by interacting with the binding sites by many drugs since the first FDA approval of the desired therapeutic effect of of endogenous small- molecule ligands such as neuro- ligand- targeting agents etanercept and infliximab in a drug. -
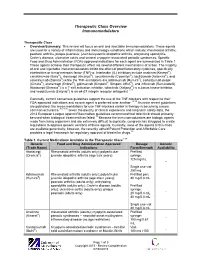
Therapeutic Class Overview Immunomodulators
Therapeutic Class Overview Immunomodulators Therapeutic Class · Overview/Summary: This review will focus on oral and injectable immunomodulators. These agents are used for a variety of inflammatory and immunologic conditions which include: rheumatoid arthritis, psoriatic arthritis, plaque psoriasis, juvenile/systemic idiopathic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis and several cryopyrin-associated periodic syndromes. Specific Food and Drug Administration (FDA)-approved indications for each agent are summarized in Table 1. These agents achieve their therapeutic effect via several different mechanisms of action. The majority of oral and injectable immunomodulators inhibit the effect of proinflammatory cytokines, specifically interleukins or tumor necrosis factor (TNF)-α. Interleukin (IL) inhibitors include anakinra (Kineret®), canakinumab (Ilaris®), rilonacept (Arcalyst®), secukinumab (Cosentyx®), tocilizumab (Actemra®), and ustekinumab (Stelara®) while the TNF-α inhibitors are adalimumab (Humira®), certolizumab pegol (Cimzia®), etanercept (Enbrel®), golimumab (Simponi®, Simponi ARIA®), and infliximab (Remicade®). Abatacept (Orencia®) is a T-cell activation inhibitor, tofacitinib (Xeljanz®) is a Janus kinase inhibitor, and vedolizumab (Entyvio®) is an α4-β7 integrin receptor antagonist.1-17 Generally, current consensus guidelines support the use of the TNF-blockers with respect to their FDA-approved indications and no one agent is preferred over another.18-35 As more recent guidelines are published, the recommendations for use TNF-blockers earlier in therapy is becoming a more common occurance.26,27,30 Given the paucity of clinical experience and long-term safety data, the 2013 European League against Rheumatism guidelines recommend that tofacitinib should primarily be used when biological treatment has failed.18 Because the immunomodulators are biologic agents made from living organisms and are extremely difficult to duplicate, congress has struggled to create regulations to approve generic versions of these agents. -

ILARIS (I-LAHR-Us) (Canakinumab) Injection for Subcutaneous Use
Medication Guide Medication Guide ILARIS (i-LAHR-us) (canakinumab) injection for subcutaneous use What is the most important information I should know about ILARIS? ILARIS can cause serious side effects, including: • Increased risk of serious infections. ILARIS can lower the ability of your immune system to fight infections. Your healthcare provider should: • test you for tuberculosis (TB) before you receive ILARIS • monitor you closely for symptoms of TB during treatment with ILARIS • check you for symptoms of any type of infection before, during, and after your treatment with ILARIS Tell your healthcare provider right away if you have any symptoms of an infection such as fever, sweats or chills, cough, flu-like symptoms, weight loss, shortness of breath, blood in your phlegm, sores on your body, warm or painful areas on your body, diarrhea or stomach pain, or feeling very tired. See "What are possible side effects of ILARIS?" for more information about side effects. What is ILARIS? ILARIS is a prescription medicine injected by your healthcare provider just below the skin (subcutaneous) used to treat: • Adults and children 4 years of age and older who have auto-inflammatory diseases called Cryopyrin-Associated Periodic Syndromes (CAPS), including: o Familial Cold Auto-inflammatory Syndrome (FCAS) o Muckle-Wells Syndrome (MWS) • Systemic Juvenile Idiopathic Arthritis (SJIA) in children 2 years of age and older. It is not known if ILARIS is safe and effective when used to treat SJIA in children under 2 years of age or when used to treat CAPS in children under 4 years of age. Who should not receive ILARIS? • Do not receive ILARIS if you are allergic to canakinumab or any of the ingredients in ILARIS. -

Secukinumab (COSENTYX) Monograph
Secukinumab (COSENTYX) Injection National Drug Monograph September 2016 VA Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives The purpose of VA PBM Services drug monographs is to provide a focused drug review for making formulary decisions. Updates will be made when new clinical data warrant additional formulary discussion. Documents will be placed in the Archive section when the information is deemed to be no longer current. FDA Approval Information Description/Mechanism of Secukinumab is a first in class recombinant, high-affinity, fully human monoclonal Action IgG1κ antibody that binds specifically to, and neutralizes the activity of, the cytokine interleukin (IL)-17A. Indication(s) Under Review Treatment of moderate to severe plaque psoriasis in adult patients who are in This Document candidates for systemic therapy or phototherapy Treatment of adult patients with active psoriatic arthritis Treatment of adult patients with active ankylosing spondylitis Dosage Form(s) Under Injection: 150 mg/mL solution in a single-use Sensoready® pen Review Injection: 150 mg/mL solution in a single-use prefilled syringe For Injection: 150 mg, lyophilized powder in a single-use vial for reconstitution for healthcare professional use only REMS REMS No REMS Postmarketing Requirements Pregnancy Rating Category B Executive Summary Efficacy Moderate to Severe Plaque Psoriasis (PPsO) Secukinumab 300 mg (SEC300, the approved dose for PPsO) showed consistently large effect sizes relative to placebo at Week 12 for at least 75% reduction in Psoriasis Area and Severity Index (PASI75), Investigator Global Assessment of 0 (clear) or 1 (almost clear) (IGA0/1) and PASI90 across four major efficacy trials (NNT range 1.3 to 1.9). -

Canakinumab (Ilaris) Effective 07/01/2021
Ilaris® (canakinumab) When requesting Ilaris® (canakinumab), the individual requiring treatment must be diagnosed with an FDA-approved indication and meet the specific coverage guidelines and applicable safety criteria for the covered indication. FDA-Approved Indications Ilaris is indicated for the treatment of: • Cryopyrin-Associated Periodic Syndromes (CAPS) including Familial Cold Autoinflammatory Syndrome, Muckle-Wells Syndrome, and Neonatal Onset Multisystem Inflammatory Disease (NOMID) or Chronic Infantile Neurological Cutaneous and Articular (CINCA) Syndrome • Tumor Necrosis Factor (TNF) Receptor Associated Periodic Syndrome (TRAPS) • Hyperimmunoglobulin D (Hyper-IgD) Syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD) • Familial Mediterranean Fever (FMF) • Active Systemic Juvenile Idiopathic Arthritis (sJIA) in patients 2 years of age and older • Adult onset Still’s Disease Coverage Guidelines The individual must meet the following criteria for initial authorization of select indications: Cryopyrin-Associated Periodic Syndromes (CAPS) • An individual is 4 years of age or older; AND • Ilaris is prescribed by or in consultation with a rheumatologist, geneticist, allergist/immunologist, or dermatologist. Familial Mediterranean Fever (FMF) • Ilaris is prescribed by or in consultation with a rheumatologist, nephrologist, geneticist, gastroenterologist, oncologist, or hematologist. Hyperimmunoglobulin D syndrome (HIDS)/Mevalonate Kinase Deficiency (MKD) OR Tumor Necrosis Factor Receptor Associated Periodic Syndrome (TRAPS) • Ilaris is