Bi-Allelic Variants in the GPI Transamidase Subunit PIGK Cause a Neurodevelopmental Syndrome with Hypotonia, Cerebellar Atrophy and Epilepsy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Innate Immune Activity Is Detected Prior to Seroconversion in Children with HLA-Conferred Type 1 Diabetes Susceptibility
2402 Diabetes Volume 63, July 2014 Henna Kallionpää,1,2,3 Laura L. Elo,1,4 Essi Laajala,1,3,5,6 Juha Mykkänen,1,3,7,8 Isis Ricaño-Ponce,9 Matti Vaarma,10 Teemu D. Laajala,1 Heikki Hyöty,11,12 Jorma Ilonen,13,14 Riitta Veijola,15 Tuula Simell,3,7 Cisca Wijmenga,9 Mikael Knip,3,16,17,18 Harri Lähdesmäki,1,3,5 Olli Simell,3,7,8 and Riitta Lahesmaa1,3 Innate Immune Activity Is Detected Prior to Seroconversion in Children With HLA-Conferred Type 1 Diabetes Susceptibility Diabetes 2014;63:2402–2414 | DOI: 10.2337/db13-1775 The insult leading to autoantibody development in chil- signatures. Genes and pathways related to innate immu- dren who will progress to develop type 1 diabetes (T1D) nity functions, such as the type 1 interferon (IFN) re- has remained elusive. To investigate the genes and sponse, were active, and IFN response factors were molecular pathways in the pathogenesis of this disease, identified as central mediators of the IFN-related tran- we performed genome-wide transcriptomics analysis on scriptional changes. Importantly, this signature was a unique series of prospective whole-blood RNA samples detected already before the T1D-associated autoanti- from at-risk children collected in the Finnish Type 1 bodies were detected. Together, these data provide Diabetes Prediction and Prevention study. We studied a unique resource for new hypotheses explaining T1D 28 autoantibody-positive children, out of which 22 pro- biology. gressed to clinical disease. Collectively, the samples PATHOPHYSIOLOGY covered the time span from before the development of autoantibodies (seroconversion) through the diagnosis of By the time of clinical diagnosis of type 1 diabetes (T1D), diabetes. -

High Mutation Frequency of the PIGA Gene in T Cells Results In
High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI A nchor−/CD52− T Cells That Can Give Early Immune Protection after This information is current as Alemtuzumab-Based T Cell−Depleted of October 1, 2021. Allogeneic Stem Cell Transplantation Floris C. Loeff, J. H. Frederik Falkenburg, Lois Hageman, Wesley Huisman, Sabrina A. J. Veld, H. M. Esther van Egmond, Marian van de Meent, Peter A. von dem Borne, Hendrik Veelken, Constantijn J. M. Halkes and Inge Jedema Downloaded from J Immunol published online 2 February 2018 http://www.jimmunol.org/content/early/2018/02/02/jimmun ol.1701018 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2018/02/02/jimmunol.170101 Material 8.DCSupplemental Why The JI? Submit online. by guest on October 1, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2018 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published February 2, 2018, doi:10.4049/jimmunol.1701018 The Journal of Immunology High Mutation Frequency of the PIGA Gene in T Cells Results in Reconstitution of GPI Anchor2/CD522 T Cells That Can Give Early Immune Protection after Alemtuzumab- Based T Cell–Depleted Allogeneic Stem Cell Transplantation Floris C. -

Complement and Inflammasome Overactivation Mediates Paroxysmal Nocturnal Hemoglobinuria with Autoinflammation
Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation Britta Höchsmann, … , Peter M. Krawitz, Taroh Kinoshita J Clin Invest. 2019. https://doi.org/10.1172/JCI123501. Research Article Hematology Inflammation Graphical abstract Find the latest version: https://jci.me/123501/pdf The Journal of Clinical Investigation RESEARCH ARTICLE Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation Britta Höchsmann,1,2 Yoshiko Murakami,3,4 Makiko Osato,3,5 Alexej Knaus,6 Michi Kawamoto,7 Norimitsu Inoue,8 Tetsuya Hirata,3 Shogo Murata,3,9 Markus Anliker,1 Thomas Eggermann,10 Marten Jäger,11 Ricarda Floettmann,11 Alexander Höllein,12 Sho Murase,7 Yasutaka Ueda,5 Jun-ichi Nishimura,5 Yuzuru Kanakura,5 Nobuo Kohara,7 Hubert Schrezenmeier,1 Peter M. Krawitz,6 and Taroh Kinoshita3,4 1Institute of Transfusion Medicine, University of Ulm, Ulm, Germany. 2Institute of Clinical Transfusion Medicine and Immunogenetics, German Red Cross Blood Transfusion Service and University Hospital Ulm, Ulm, Germany. 3Research Institute for Microbial Diseases and 4WPI Immunology Frontier Research Center, Osaka University, Osaka, Japan. 5Department of Hematology and Oncology, Graduate School of Medicine, Osaka University, Osaka, Japan. 6Institute for Genomic Statistics and Bioinformatics, Rheinische Friedrich-Wilhelms-Universität Bonn, Bonn, Germany. 7Department of Neurology, Kobe City Medical Center General Hospital, Kobe, Japan. 8Department of Tumor Immunology, Osaka -

Profiling the Expression Pattern of GPI Transamidase Complex Subunits in Human Cancer
Modern Pathology (2008) 21, 979–991 & 2008 USCAP, Inc All rights reserved 0893-3952/08 $30.00 www.modernpathology.org Profiling the expression pattern of GPI transamidase complex subunits in human cancer Jatin K Nagpal1,5, Santanu Dasgupta1,5, Sana Jadallah2, Young K Chae1, Edward A Ratovitski3, Antoun Toubaji2, George J Netto2, Toby Eagle1, Aviram Nissan4, David Sidransky1 and Barry Trink1 1Department of Otolaryngology-Head and Neck Surgery, Head and Neck Cancer Research Division, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 2Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 3Department of Dermatology, Johns Hopkins University School of Medicine, Baltimore, MD, USA and 4Department of Surgery, Hadassah University Hospital, Mount Scopus, Jerusalem, Israel The glycosylphosphatidylinositol transamidase complex (GPIT) consists of five subunits: PIG-U, PIG-T, GPAA1, PIG-S and GPI8, and is important in attaching GPI anchors to target proteins. On the basis of our previous reports incriminating PIG-U as an oncogene in bladder cancer and PIG-T and GPAA1 as oncogenes in breast cancer, we evaluated the expression pattern of the GPIT subunits in 19 different human cancers at both mRNA and protein levels. In general, our results demonstrate a more frequent expression of GPIT subunits in cancers than in normal. Among the 19 anatomic sites compared; breast, ovary and uterus showed consistent evidence of overexpression of specific GPIT subunits. There was also overexpression of PIG-U and GPI8 in lymphoma. In addition, non-small cell lung carcinoma showed significant overexpression of the GPIT subunits as compared to small cell lung carcinoma and normal lung tissue. -

Supplemental Figures 04 12 2017
Jung et al. 1 SUPPLEMENTAL FIGURES 2 3 Supplemental Figure 1. Clinical relevance of natural product methyltransferases (NPMTs) in brain disorders. (A) 4 Table summarizing characteristics of 11 NPMTs using data derived from the TCGA GBM and Rembrandt datasets for 5 relative expression levels and survival. In addition, published studies of the 11 NPMTs are summarized. (B) The 1 Jung et al. 6 expression levels of 10 NPMTs in glioblastoma versus non‐tumor brain are displayed in a heatmap, ranked by 7 significance and expression levels. *, p<0.05; **, p<0.01; ***, p<0.001. 8 2 Jung et al. 9 10 Supplemental Figure 2. Anatomical distribution of methyltransferase and metabolic signatures within 11 glioblastomas. The Ivy GAP dataset was downloaded and interrogated by histological structure for NNMT, NAMPT, 12 DNMT mRNA expression and selected gene expression signatures. The results are displayed on a heatmap. The 13 sample size of each histological region as indicated on the figure. 14 3 Jung et al. 15 16 Supplemental Figure 3. Altered expression of nicotinamide and nicotinate metabolism‐related enzymes in 17 glioblastoma. (A) Heatmap (fold change of expression) of whole 25 enzymes in the KEGG nicotinate and 18 nicotinamide metabolism gene set were analyzed in indicated glioblastoma expression datasets with Oncomine. 4 Jung et al. 19 Color bar intensity indicates percentile of fold change in glioblastoma relative to normal brain. (B) Nicotinamide and 20 nicotinate and methionine salvage pathways are displayed with the relative expression levels in glioblastoma 21 specimens in the TCGA GBM dataset indicated. 22 5 Jung et al. 23 24 Supplementary Figure 4. -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -
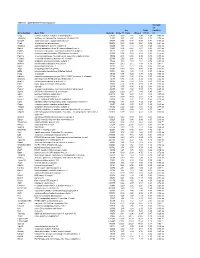
Table S-1 Mpkccd Transcriptome
Table S1. mpkCCD Cell Transcriptomes. Correlati on Ori. Ratio Coefficie Gene Symbol Gene Title GeneID Clone 11 Clone Clone 2 (11/2) nt Copg coatomer protein complex, subunit gamma 54161 1.99 1.81 2.48 0.80 -0.49 ns Atp6v0d1 ATPase, H+ transporting, lysosomal V0 subunit D1 11972 3.37 2.87 3.00 1.12 0.74 ns Golga7 golgi autoantigen, golgin subfamily a, 7 57437 3.03 3.40 3.58 0.84 -0.34 ns Psph phosphoserine phosphatase 100678 0.38 0.66 0.64 0.59 -0.61 ns Trappc4 trafficking protein particle complex 4 60409 1.17 1.12 1.08 1.08 0.64 ns Dpm2 dolichol-phosphate (beta-D) mannosyltransferase 2 13481 0.74 0.87 0.81 0.91 -0.31 ns Psmb5 proteasome (prosome, macropain) subunit, beta type 5 19173 2.92 3.00 2.92 0.99 0.32 ns Dhrs1 dehydrogenase/reductase (SDR family) member 1 52585 0.74 0.68 0.88 0.83 -0.18 ns Ppm1a protein phosphatase 1A, magnesium dependent, alpha isoform 19042 2.43 2.60 2.96 0.82 -0.53 ns Psenen presenilin enhancer 2 homolog (C. elegans) 66340 2.52 2.88 3.80 0.66 -0.77 ns Anapc1 anaphase promoting complex subunit 1 17222 1.16 1.33 1.61 0.72 -0.33 ns Mrpl43 mitochondrial ribosomal protein L43 94067 2.15 2.15 1.84 1.16 0.91 * Nmt1 N-myristoyltransferase 1 18107 3.21 2.71 3.36 0.95 0.31 ns Atg5 autophagy-related 5 (yeast) 11793 0.70 0.73 0.66 1.05 -0.23 ns Mtif2 mitochondrial translational initiation factor 2 76784 1.16 1.29 1.19 0.97 -0.36 ns Psap prosaposin 19156 8.07 7.25 8.37 0.96 0.02 ns Ube2g1 ubiquitin-conjugating enzyme E2G 1 (UBC7 homolog, C. -

The Glycosylphosphatidylinositol Biosynthesis Pathway in Human Diseases Tenghui Wu1,2, Fei Yin1,2, Shiqi Guang1,2, Fang He1,2, Li Yang1,2 and Jing Peng1,2*
Wu et al. Orphanet Journal of Rare Diseases (2020) 15:129 https://doi.org/10.1186/s13023-020-01401-z REVIEW Open Access The Glycosylphosphatidylinositol biosynthesis pathway in human diseases Tenghui Wu1,2, Fei Yin1,2, Shiqi Guang1,2, Fang He1,2, Li Yang1,2 and Jing Peng1,2* Abstract Glycosylphosphatidylinositol biosynthesis defects cause rare genetic disorders characterised by developmental delay/ intellectual disability, seizures, dysmorphic features, and diverse congenital anomalies associated with a wide range of additional features (hypotonia, hearing loss, elevated alkaline phosphatase, and several other features). Glycosylphosphatidylinositol functions as an anchor to link cell membranes and protein. These proteins function as enzymes, adhesion molecules, complement regulators, or co-receptors in signal transduction pathways. Biallelic variants involved in the glycosylphosphatidylinositol anchored proteins biosynthetic pathway are responsible for a growing number of disorders, including multiple congenital anomalies-hypotonia-seizures syndrome; hyperphosphatasia with mental retardation syndrome/Mabry syndrome; coloboma, congenital heart disease, ichthyosiform dermatosis, mental retardation, and ear anomalies/epilepsy syndrome; and early infantile epileptic encephalopathy-55. This review focuses on the current understanding of Glycosylphosphatidylinositol biosynthesis defects and the associated genes to further understand its wide phenotype spectrum. Keywords: GPI-APs, PIG/PGAP genes, Phenotype Introduction can be divided into the -

Chipenrich: a Package for Gene Set Enrichment Testing of Genome Region Data
chipenrich: A package for gene set enrichment testing of genome region data R.P. Welch, C. Lee, R.A. Smith, P. Imbriano, S. Patil, T. Weymouth, L.J. Scott, M.A. Sartor October 14, 2013 Contents 1 Introduction 1 2 Synopsis 1 3 Locus definitions 3 4 Mappability 3 4.1 Base pair mappability . 3 4.2 Locus mappability . 4 5 Examples 4 5.1 Diagnostic plots . 4 5.2 ChIP-Enrich . 4 5.3 ChIP-Enrich with mappability . 4 5.4 Fisher's exact test . 8 6 Output 9 6.1 Peak-to-gene assignments . 9 6.2 Gene set enrichment test results . 9 7 References 10 1 Introduction This document describes how to use chipenrich to analyze results from ChIP-Seq experiments and other DNA sequencing experiments that result in a set of genomic regions. chipenrich includes a method that adjusts for potential confounders of gene set enrichment testing (locus length and mappability of the sequence reads.) 2 Synopsis After starting R, the package should be loaded using the following: > library(chipenrich) This will load chipenrich, the chipenrich.data package, as well as all other dependency packages. The main function for conducting all gene set enrichment testing is chipenrich(). The defaults for the chipenrich() function are chipenrich(peaks, out_name = "chipenrich", out_path = getwd(), genome = "hg19", genesets = ('GOBP', 'GOCC', 'GOMF'), locusdef = "nearest_tss ", method = "chipenrich", fisher_alt = "two.sided", use_mappability = F, read_length = 24, genome_length = NULL, qc_plots = T) 1 The \peaks" option should be either a data frame or character vector representing the path to a file containing the peaks. The file (or data frame) should have at least 3 columns: \chrom," \start," and \end," denoting the chromosome, starting position, and ending position of the peak. -

Ethanolamine Phosphate on the Second Mannose As Bridge in GPI Anchored Proteins
bioRxiv preprint doi: https://doi.org/10.1101/2020.11.26.399477; this version posted November 26, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Ethanolamine phosphate on the second mannose as bridge in GPI anchored proteins: Towards understanding inherited PIGG deficiency Mizuki Ishida1, Yuta Maki2,3, Akinori Ninomiya4, Yoko Takada5, Philippe M Campeau6, Taroh Kinoshita1,5, Yoshiko Murakami1,5* 1Yabumoto Department of Intractable Disease Research, Research Institute for Microbial Diseases, Osaka University, Suita, Osaka, Japan 565-0871; 2Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka, Japan 560-0043; 3Project Research Center for Fundamental Sciences, Graduate School of Science, Osaka University, Toyonaka, Osaka, Japan 560-0043; 4Central Instrumentation Laboratory, Research Institute for Microbial Diseases, Osaka University, Suita, Osaka, Japan 565-0871; 5WPI Immunology Frontier Research Center, Osaka University, Suita, Osaka, Japan 565-0871; 6Department of Pediatrics, CHU Sainte-Justine and University of Montreal, Montreal, QC, Canada, H3T 1C5 A corresponding author: Yoshiko Murakami MD. PhD. Yabumoto Department of Intractable Disease Research Research Institute for Microbial Diseases, Osaka University 3-1 Yamada-oka, Suita, Osaka 565-0871, Japan [email protected] tel +81-6-6879-8329 fax +81-6-6875-5233 Running title: Novel structure of GPI anchored proteins Keywords GPI transamidase, Mass spectrometry, PI-PLC 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.11.26.399477; this version posted November 26, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Simulation and Estimation of Gene Number in a Biological Pathway
Tokunaga et al. BMC Genomics 2014, 15:1016 http://www.biomedcentral.com/1471-2164/15/1016 RESEARCH ARTICLE Open Access Simulation and estimation of gene number in a biological pathway using almost complete saturation mutagenesis screening of haploid mouse cells Masahiro Tokunaga1, Chikara Kokubu1, Yusuke Maeda2,3, Jun Sese4, Kyoji Horie1,7, Nakaba Sugimoto5, Taroh Kinoshita2,3, Kosuke Yusa6* and Junji Takeda1* Abstract Background: Genome-wide saturation mutagenesis and subsequent phenotype-driven screening has been central to a comprehensive understanding of complex biological processes in classical model organisms such as flies, nematodes, and plants. The degree of “saturation” (i.e., the fraction of possible target genes identified) has been shown to be a critical parameter in determining all relevant genes involved in a biological function, without prior knowledge of their products. In mammalian model systems, however, the relatively large scale and labor intensity of experiments have hampered the achievement of actual saturation mutagenesis, especially for recessive traits that require biallelic mutations to manifest detectable phenotypes. Results: By exploiting the recently established haploid mouse embryonic stem cells (ESCs), we present an implementation of almost complete saturation mutagenesis in a mammalian system. The haploid ESCs were mutagenized with the chemical mutagen N-ethyl-N-nitrosourea (ENU) and processed for the screening of mutants defective in various steps of the glycosylphosphatidylinositol-anchor biosynthetic pathway. The resulting 114 independent mutant clones were characterized by a functional complementation assay, and were shown to be defective in any of 20 genes among all 22 known genes essential for this well-characterized pathway. Ten mutants were further validated by whole-exome sequencing.