Study Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accidental Vaccinia Virus Exposure: Information Sheet for Laboratory Workers
The University of Iowa Vaccinia Vaccination Information Form I understand that due to my occupational exposure to vaccinia virus, I may be at risk of acquiring infection from vaccinia. I have been given the opportunity to be vaccinated with the smallpox vaccine, at no charge to myself. After reading the Medication Guide and information packet from the Centers for Disease Control and Prevention (CDC) I understand there are risks involved in working with vaccinia virus, in addition to risks in receving the vaccination. My signature below indicates that I have: 1. Read the CDC’s Medication Guide that has been provided to me (follows this informational form). 2. Read the CDC’s "Important Information about Vaccinia (Smallpox) Vaccine For Laboratory Workers" that has been provided to me (follows this informational form). 3. Had an opportunity to ask questions about vaccinia with Biosafety Staff in the Environmental Health and Safety Office (EHS) at 335-8501. 4. Had the opportunity to ask questions about the vaccine with University Employee Health Clinic staff at 356-3631. I have decided to: Decline the vaccination at this time. I understand that by declining this vaccine, I continue to be at risk of acquiring an infection from vaccinia. If I continue to have occupational exposure to this virus and decide to be vaccinated in the future, I may request and receive the vaccination, at no charge to myself. Receive the vaccine, provided through the University Employee Health Clinic (UEHC). Signature: Please print name: Date: University ID number: Principal Investigator/Lab Director Name: If consenting, please also complete: Date of Birth: Job Title: List of duties and/or viruses used in lab that may cause exposure to vaccinia virus: Return page 1 of this form to: Associate Biosafety Officer, 100 EHS. -

ACAM2000 Clonal Vero Cell Culture Vaccinia Virus (New York City Board of Health Strain) — a Second-Generation Smallpox Vaccine for Biological Defense
International Journal of Infectious Diseases (2004) 8S2, S31—S44 http://intl.elsevierhealth.com/journals/ijid ACAM2000 clonal Vero cell culture vaccinia virus (New York City Board of Health strain) — a second-generation smallpox vaccine for biological defense Thomas P. Monatha,*, Joseph R. Caldwella, Wolfgang Mundtb, Joan Fuscob, Casey S. Johnsonc, Mark Bullerd, Jian Liua, Bridget Gardnera, Greg Downinga, Paul S. Bluma, Tracy Kempa, Richard Nicholsa, Richard Weltzina aAcambis Inc., 38, Sidney Street, Cambridge, MA 02139, USA bBaxter BioScience, USA and Austria cPRA International, Lenexa, KS dSt. Louis University Medical School, St. Louis MO Summary The threat of smallpox as a biological weapon has spurred efforts to create stockpiles of vaccine for emergency preparedness. In lieu of preparing vaccine in animal skin (the original method), we cloned vaccinia virus (New York City Board of Health strain, Dryvax1) by plaque purification and amplified the clone in cell culture. The overarching goal was to produce a modern vaccine that was equivalent to the currently licensed Dryvax1 in its preclinical and clinical properties, and could thus reliably protect humans against smallpox. A variety of clones were evaluated, and many were unacceptably virulent in animal models. One clonal virus (ACAM1000) was selected and produced at clinical grade in MRC-5 human diploid cells. ACAM1000 was comparable to Dryvax1 in immunogenicity and protective activity but was less neurovirulent for mice and nonhuman primates. To meet requirements for large quantities of vaccine after the events of September 11th 2001, the ACAM1000 master virus seed was used to prepare vaccine (designated ACAM2000) at large scale in Vero cells under serum-free conditions. -

IMVANEX, Common Name – Modified Vaccinia Ankara Virus
ANNEX I SUMMARY OF PRODUCT CHARACTERISTICS 1 This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions. 1. NAME OF THE MEDICINAL PRODUCT IMVANEX suspension for injection Smallpox vaccine (Live Modified Vaccinia Virus Ankara) 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One dose (0.5 ml) contains: Modified Vaccinia Ankara – Bavarian Nordic Live virus1 no less than 5 x 107 Inf.U * *infectious units 1 Produced in chick embryo cells This vaccine contains trace residues of chicken protein, benzonase, gentamicin and ciprofloxacin (see section 4.3). For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Suspension for injection. Light yellow to pale white, milky suspension. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Active immunisation against smallpox in adults (see sections 4.4 and 5.1). The use of this vaccine should be in accordance with official recommendations. 4.2 Posology and method of administration Posology Primary vaccination (individuals previously not vaccinated against smallpox): A first dose of 0.5 ml should be administered on an elected date. A second dose of 0.5 ml should be administered no less than 28 days after the first dose. See sections 4.4 and 5.1. Booster vaccination (individuals previously vaccinated against smallpox): There are inadequate data to determine the appropriate timing of booster doses. If a booster dose is considered necessary then a single dose of 0.5 ml should be administered. See sections 4.4 and 5.1. -

IMVAMUNE® and ACAM2000® Provide Different Protection Against
Article IMVAMUNE® and ACAM2000® Provide Different Protection against Disease When Administered Postexposure in an Intranasal Monkeypox Challenge Prairie Dog Model M. Shannon Keckler 1,2 , Johanna S Salzer 1,3, Nishi Patel 1,4, Michael B Townsend 1, Yoshinori J Nakazawa 1, Jeffrey B Doty 1, Nadia F Gallardo-Romero 1, Panayampalli S Satheshkumar 1, Darin S Carroll 1,5, Kevin L Karem 1,6 and Inger K Damon 1,* 1 Poxvirus and Rabies Branch, Division of High-Consequence Pathogens and Pathology, Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, Atlanta, GA 30333, USA; [email protected] (M.S.K.); [email protected] (J.S.S.); [email protected] (N.P.); [email protected] (M.B.T.); [email protected] (Y.J.N.); [email protected] (J.B.D.); [email protected] (N.F.G.-R.); [email protected] (P.S.S.); [email protected] (D.S.C.); [email protected] (K.L.K.) 2 Clinical and Environmental Microbiology Branch, Division of Healthcare Quality Promotion, Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, Atlanta, GA 30333, USA 3 Bacterial Special Pathogens Branch, Division of High-Consequence Pathogens and Pathology, Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, Atlanta, GA 30333, USA 4 Respiratory Diseases Branch, Division of Bacterial Diseases, Centers for Disease Control and Prevention, U.S. Department of Health and Human Services, Atlanta, GA 30333, USA 5 Office of the Director, National Center for Emerging and Zoonotic Diseases, Centers for Disease Control and Prevention, U.S. -

Guidelines for Vaccination of Adult Solid Organ Transplant Candidates and Recipients
Stanford Healthcare Vaccination Subcommittee Issue Date: 7/2018 Guidelines for Vaccination of Adult Solid Organ Transplant Candidates and Recipients A. General considerations regarding vaccination 1. Adult solid organ transplant (SOT) candidates and recipients should receive all vaccines indicated based on their ages, medical conditions, and other factors that apply to non- SOT candidates or recipients (see http://www.cdc.gov/vaccines/schedules/hcp/adult.html, Appendix A, and Appendix B) except for the below-listed exceptions or additions. • All SOT candidates and recipients should be vaccinated against pneumococcus with PCV13 and PPSV23. • All SOT candidates and recipients should receive a HepB vaccine series with post-vaccination titers unless they have a documented anti-HBs titer of ≥ 10 mIU/mL after a properly-timed HepB series or unless they have known hepatitis B virus infection. • All SOT candidates and recipients should receive a HepA vaccine series unless previously administered or unless they have a positive hepatitis A virus immunoglobulin G assay. • Live-attenuated vaccines1 should not be administered to SOT recipients, SOT candidates on immunosuppression, or SOT candidates who may undergo SOT within 4 weeks. The timing of inactivated, subunit, or toxoid vaccines is discussed below. Vaccine Notes Influenza Can be given pre- and/or post-SOT (see Appendix C) Tdap or Td Can be given pre- and/or post-SOT Live-attenuated vaccine to be given pre-SOT only; generally those born MMR before 1957 (among others) are considered immune -

(GRADE) for Use of Smallpox Vaccine in Laboratory and Health
Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) for Use of Smallpox Vaccine in Laboratory and Health-Care Personnel at Risk for Occupational Exposure to Orthopoxviruses: Recommendations of the Advisory Committee on Immunization Practices, 2015. Background ACAM2000, a live vaccinia virus vaccine, is the only smallpox vaccine currently licensed and available in the United States (U.S.) for vaccination of persons at risk for orthopoxviral disease. ACAM2000 is produced in Vero cells and derived from a clonal isolate of Dryvax, the New York City Board of Health strain widely used during the smallpox eradication campaign [1-5]. Like Dryvax, ACAM2000 is administered percutaneously using a bifurcated needle, and comes with potential risks of serious adverse events [6-7]. Recommendations of the U.S. Advisory Committee on Immunization Practices (ACIP) regarding smallpox (vaccinia) vaccination, most recently revised in 2003, specify Dryvax as the designated smallpox vaccine in routine non-emergency vaccination programs, which primarily involve laboratory and health-care personnel, as well as select military personnel [8-9]. However, as the license for Dryvax was withdrawn in 2008, and remaining vaccine supplies were subsequently destroyed, the need to develop new ACIP recommendations based on ACAM2000 is paramount [10]. Thus, the ACIP Smallpox Vaccine Work Group has applied the GRADE framework to the available evidence evaluating the administration of ACAM2000 in laboratory and health-care personnel at risk for orthopoxviral -

Nebraska State Immunization Information System – Flat File Specification Version 10.1.0 (Revised 7/09/2021)
Nebraska State Immunization Information System – Flat File Specification Version 10.1.0 (Revised 7/09/2021) Immunization data is passed to the central registry using three flat files containing client, immunization, and comment information (optional) respectively. The files will be linked via a 24-character Record Identifier supplied by the provider of the file. This identifier will uniquely identify each client and will appear in each immunization and comment (optional) record to link the immunization and comment (optional) to the client. Character fields need to be left justified and blank-filled, number fields right justified and blank- filled, and date fields in format MMDDYYYY with leading zeroes. If a site is unable to supply any information for a specified field, the entire field needs be filled with spaces. Below are the fields to include in each of the files. Files need to be generated using the ASCII character set. Records will be fixed length and need to be terminated with a carriage return/line feed. If you have questions on concerning flat files, contact the Help Desk at [email protected]. Client Data Column Data type Required Default Notes Record Identifier Char(24) Y Supplied by sender, used to link a Client to Immunization records. Client Status Char(1) A Use the NESIIS code set for Client Status. First Name Char(25) Y If client does not have a first name, NO FIRST NAME must be entered in this field. Middle Name Char(25) Last Name Char(35) Y Name Suffix Char(10) JR, III, etc. Birth Date Date(8) Y MMDDYYYY Death Date Date(8) MMDDYYYY Mothers First Name Char(25) These are mandatory fields in NESIIS. -

Cross-Neutralizing and Protective Human Antibody Specificities To
Article Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections Graphical Abstract Authors Iuliia Gilchuk, Pavlo Gilchuk, Gopal Sapparapu, ..., Gary H. Cohen, Sebastian Joyce, James E. Crowe, Jr. Correspondence [email protected] In Brief Protective immunity against smallpox and other members of the orthopoxvirus family requires the cooperation of antibodies that target different viral proteins at distinct stages of maturation of the virus. Highlights d Orthopoxviruses elicit a complex B cell immune response reactive to diverse antigens d A large fraction of orthopoxvirus neutralizing mAbs possess cross-neutralizing activity d Six principal mAb specificities participate in cross- neutralization and protection d Most efficient protection is achieved by mixture of diverse mAbs specificities Gilchuk et al., 2016, Cell 167, 684–694 October 20, 2016 ª 2016 Elsevier Inc. http://dx.doi.org/10.1016/j.cell.2016.09.049 Article Cross-Neutralizing and Protective Human Antibody Specificities to Poxvirus Infections Iuliia Gilchuk,1 Pavlo Gilchuk,2,3 Gopal Sapparapu,1,4 Rebecca Lampley,1 Vidisha Singh,1 Nurgun Kose,1 David L. Blum,1 Laura J. Hughes,5 Panayampalli S. Satheshkumar,5 Michael B. Townsend,5 Ashley V. Kondas,5 Zachary Reed,5,6 Zachary Weiner,6 Victoria A. Olson,5 Erika Hammarlund,7 Hans-Peter Raue,7 Mark K. Slifka,7 James C. Slaughter,8 Barney S. Graham,9 Kathryn M. Edwards,4 Roselyn J. Eisenberg,10 Gary H. Cohen,11 Sebastian Joyce,2,3 and James E. Crowe, Jr.1,2,4,12,* 1The Vanderbilt Vaccine Center, Vanderbilt -

US Vaccine Table
United States Vaccine Names United States Vaccines Doses in Routine Vaccine Trade Name Abbreviation Manufacturer Route Approved Ages Comments Series Adenovirus N/A Teva Oral 1 17-50 years Live: Approved for military Pharmaceutical populations; not approved Adenovirus Type 4 & Type 7 (2 Tablets) Industries Ltd. for pregnant women BioThrax® AVA Emergent IM 3 18-65 years Cell-free filtrate from Anthrax BioSolutions avirulent strain, Adj. Vaxchora™† N/A Emergent Oral (Liquid) 1 18-64 years Live Attenuated Cholera BioSolutions Daptacel® DTaP Sanofi IM 5 6 weeks-6 years Inactivated, Adj. DTaP Infanrix™ DTaP GlaxoSmithKline IM 5 6 weeks-6 years Inactivated, Adj. N/A (Generic) DT Sanofi IM 5 6 weeks-6 years Inactivated, Adj.: Use DT when pertussis is contraindicated ActHIB® Hib (PRP-T) Sanofi IM 4 2 months-5 years Inactivated (Tetanus toxoid conjugate) Haemophilus Hiberix™ Hib (PRP-T) GlaxoSmithKline IM 4 6 weeks- 4 years Inactivated (Tetanus toxoid influenzae type b (Hib) conjugate) PedvaxHIB® Hib (PRP-OMP) Merck IM 3 2-71 months Inactivated, Adj. (Meningococcal conjugate) Havrix™ HepA GlaxoSmithKline IM 2 Pediatric: Inactivated, Adj. 12 months-18 years; Adult: ≥19 years Hepatitis A Vaqta® HepA Merck IM 2 Pediatric: Inactivated, Adj. 12 months-18 years; Adult: ≥19 years Engerix-B™ HepB GlaxoSmithKline IM 3 Pediatric: Recombinant, Adj. Birth-19 years Adult: ≥20 years Recombivax HB® HepB Merck IM 3 Pediatric: Recombinant, Adj. Hepatitis B Appendix B Birth-19 years Adult: ≥20 years Appendix B-3 Heplisav-B® HepB Dynavax IM 2 ≥18 years Recombinant, Adj. Technologies Herpes Zoster Shingrix™ RZV GlaxoSmithKline IM 2 ≥50 years Recombinant, Adj. -

VALID VALUES Last Updated 3/4/20
FLORIDA SHOTS VALID VALUES Last Updated 3/4/20 CVX Codes See also: http://www2a.cdc.gov/vaccines/IIS/IISStandards/vaccines.asp?rpt=cvx Code Description FLSHOTS Vacc. Type 1 Diphtheria, tetanus toxoids and pertussis DTP 2 Trivalent poliovirus, live, oral OPV 3 Measles, mumps, and rubella virus MMR 4 Measles and rubella virus MR 5 Measles virus MEASLES 6 Rubella virus RUBELLA 7 Mumps virus MUMPS 8 Hepatitis B, pediatric or pediatric/adolescent HEP B PED 9 Tetanus and diphtheria toxoids, adsorbed, pf, adult TD PF (TDVAX) 10 Poliovirus, inactivated IPV 11 Pertussis PERTUSSIS 12 Diphtheria antitoxin DIPHTHERIA ANTITOXIN 13 Tetanus immune globulin TIG 14 Immune globulin, unspecified IG UNK 17 Haemophilus influenza type B, unspecified HIB UNK 19 Bacillus Calmette-Guerin BCG 20 Diphtheria, tetanus toxoids and acellular pertussis DTAP 21 Varicella virus VZV 22 DTP-Haemophilus influenzae type b conjugate DTP-HIB 23 Plague PLAGUE 24 Anthrax ANTHRAX 25 Typhoid, live, oral TYPHOID PO 26 Cholera, unspecified CHOLERA UNK 27 Botulinum antitoxin BOTULINUM 28 Diphtheria and tetanus toxoids, adsorbed, pediatric DT 29 Cytomagalovirus IG IV CMVIG 30 Hepatitis B immune globulin HBIG 32 Meningococcal polysaccharide MPSV4 33 Pneumoccocal polysaccharide PPSV23 34 Rabies immune globulin RIG 35 Tetanus toxoid, adsorbed TT 36 Varicella zoster immune globulin VZIG 37 Yellow fever (YF-VAX) YELLOW FEVER (YF-VAX) 38 Rubella and mumps virus MUMPS-RUB 39 Japanese encephalitis, SC JENCEPH SC 40 Rabies, intradermal RABIES ID 41 Typhoid, parenteral, non-AKD TYPHOID PAR 42 Hepatitis B, adol/high risk infant HEP B HIGH RISK 43 Hepatitis B, adult HEP B ADULT 44 Hepatitis B, dialysis HEP B DIALYSIS 45 Hepatitis B, unspecified HEP B UNK 46 Haemophilus influenza type B, PRP-D HIB PRP-D 47 Haemophilus influenza type B, HbOC HIB HBOC 48 Haemophilus influenza type B, PRP-T HIB PRP-T 1 FLORIDA SHOTS VALID VALUES Last Updated 3/4/20 Code Description FLSHOTS Vacc. -

Supported MCIR Vaccine Codes Including U.S. Licensed CVX, CPT
UPDATE: August 3, 2021 includes COVID-19 Supported MCIR Vaccine Codes Including U.S. Licensed CVX, CPT-4, and MVX MCIR codes are a reflection, but not a complete list of, those maintained at the CDC National Immunization Program Website. For additional IIS Vaccine Code information please visit CDC’s IIS NDC Lookup Crosswalk Table. CDC Current CDC MCIR Vaccine Display Active/ CDC Product Procedural Manufacturer Vaccine CDC Vaccine Name Manufacturer Name Historical Name Terminology Code (MVX) Code (CVX) Code (CPT) Emergent 24 Anthrax Active BIOTHRAX Anthrax 90581 Biosolutions MIP cholera, live 174 Cholera (Vaxchora) Active attenuated Vaxchora 90625 PaxVax PAX COVID-19, mRNA, LNP-S, PF, 207 COVID-19 (Moderna) Active COVID-19 100 mcg/ 0.5 mL dose 91301 Moderna US, Inc MOD COVID-19, mRNA, LNP-S, PF, 208 COVID-19 (Pfizer) Active COVID-19 30 mcg/0.3 mL dose 91300 Pfizer-BioNTech PFR COVID-19 vaccine, vector-nr, 212 COVID-19 (Janssen) Active COVID-19 rS-Ad26, PF, 0.5 mL 91303 Janssen Products, LP JSN Diphtheria-Tetanus (DT 28 DT (pediatric) Active DT (GENERIC) pediatric) 90702 Sanofi PMC 106 DTaP Daptacel Active DAPTACEL DTaP (5 pertussis) 90700 Sanofi PMC 20 DTaP (Ped) Infanrix Active INFANRIX DTaP 90700 GlaxoSmithKline SKB DTaP-HepB-IPV 110 (Pediarix) Active PEDIARIX DTaP-Hep B-IPV 90723 GlaxoSmithKline SKB DTaP-Hib-IPV 120 (Pentacel) Active PENTACEL DTaP-Hib-IPV 90698 Sanofi PMC MSP Vaccine 146 DTaP-IPV-Hib-HepB Active VAXELIS DTaP-IPV-Hib-HepB 90697 Co(partnership Merck MSP and SanofiPasteur 130 DTaP-IPV Active KINRIX DTaP-IPV 90696 GlaxoSmithKline -
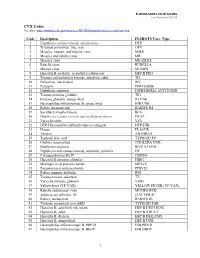
VALID VALUES Last Updated 8/20/18
FLORIDA SHOTS VALID VALUES Last Updated 8/20/18 CVX Codes See also: http://www2a.cdc.gov/vaccines/IIS/IISStandards/vaccines.asp?rpt=cvx Code Description FLSHOTS Vacc. Type 1 Diphtheria, tetanus toxoids and pertussis DTP 2 Trivalent poliovirus, live, oral OPV 3 Measles, mumps, and rubella virus MMR 4 Measles and rubella virus MR 5 Measles virus MEASLES 6 Rubella virus RUBELLA 7 Mumps virus MUMPS 8 Hepatitis B, pediatric or pediatric/adolescent HEP B PED 9 Tetanus and diphtheria toxoids, adsorbed, adult TD 10 Poliovirus, inactivated IPV 11 Pertussis PERTUSSIS 12 Diphtheria antitoxin DIPHTHERIA ANTITOXIN 13 Tetanus immune globulin TIG 14 Immune globulin, unspecified IG UNK 17 Haemophilus influenza type B, unspecified HIB UNK 18 Rabies intramuscular RABIES IM 19 Bacillus Calmette-Guerin BCG 20 Diphtheria, tetanus toxoids and acellular pertussis DTAP 21 Varicella virus VZV 22 DTP-Haemophilus influenza type b conjugate DTP-HIB 23 Plague PLAGUE 24 Anthrax ANTHRAX 25 Typhoid, live, oral TYPHOID PO 26 Cholera, unspecified CHOLERA UNK 27 Botulinum antitoxin BOTULINUM 28 Diphtheria and tetanus toxoids, adsorbed, pediatric DT 29 Cytomagalovirus IG IV CMVIG 30 Hepatitis B immune globulin HBIG 32 Meningococcal polysaccharide MPSV4 33 Pneumoccocal polysaccharide PPSV23 34 Rabies immune globulin RIG 35 Tetanus toxoid, adsorbed TT 36 Varicella immune globulin VZIG 37 Yellow fever (YF-VAX) YELLOW FEVER (YF-VAX) 38 Rubella and mumps virus MUMPS-RUB 39 Japanese encephalitis, SC JENCEPH SC 40 Rabies, intradermal RABIES ID 41 Typhoid, parenteral, non-AKD TYPHOID