7-ACVR1.1.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-Müllerian Hormone in Stallions and Mares: Physiological Variations, Clinical Applications, and Molecular Aspects
University of Kentucky UKnowledge Theses and Dissertations--Veterinary Science Veterinary Science 2014 ANTI-MÜLLERIAN HORMONE IN STALLIONS AND MARES: PHYSIOLOGICAL VARIATIONS, CLINICAL APPLICATIONS, AND MOLECULAR ASPECTS Anthony N.J. Claes University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Claes, Anthony N.J., "ANTI-MÜLLERIAN HORMONE IN STALLIONS AND MARES: PHYSIOLOGICAL VARIATIONS, CLINICAL APPLICATIONS, AND MOLECULAR ASPECTS" (2014). Theses and Dissertations-- Veterinary Science. 18. https://uknowledge.uky.edu/gluck_etds/18 This Doctoral Dissertation is brought to you for free and open access by the Veterinary Science at UKnowledge. It has been accepted for inclusion in Theses and Dissertations--Veterinary Science by an authorized administrator of UKnowledge. For more information, please contact [email protected]. STUDENT AGREEMENT: I represent that my thesis or dissertation and abstract are my original work. Proper attribution has been given to all outside sources. I understand that I am solely responsible for obtaining any needed copyright permissions. I have obtained needed written permission statement(s) from the owner(s) of each third-party copyrighted matter to be included in my work, allowing electronic distribution (if such use is not permitted by the fair use doctrine) which will be submitted to UKnowledge as Additional File. I hereby grant to The University of Kentucky and its agents the irrevocable, non-exclusive, and royalty-free license to archive and make accessible my work in whole or in part in all forms of media, now or hereafter known. I agree that the document mentioned above may be made available immediately for worldwide access unless an embargo applies. -

A Meta-Analysis of the Inhibin Network Reveals Prognostic Value in Multiple Solid Tumors
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.25.171942; this version posted June 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 A meta-analysis of the inhibin network reveals prognostic value in multiple solid tumors Eduardo Listik1†, Ben Horst1,2†, Alex Seok Choi1, Nam. Y. Lee3, Balázs Győrffy4 and Karthikeyan Mythreye1 Affiliations: 1Department of Pathology, University of Alabama at Birmingham, Birmingham AL, USA, 35294. 2Department of Chemistry and Biochemistry, University of South Carolina, Columbia SC, USA, 29208. 3 Division of Pharmacology, Chemistry and Biochemistry, College of Medicine, University of Arizona, Tucson, AZ, 85721, USA. 4 TTK Cancer Biomarker Research Group, Institute of Enzymology, and Semmelweis University Department of Bioinformatics and 2nd Department of Pediatrics, Budapest, Hungary. Corresponding author: Karthikeyan Mythreye, Ph.D. Division of Molecular and Cellular Pathology, Department of Pathology, The University of Alabama at Birmingham. WTI 320B, 1824 Sixth Avenue South, Birmingham, AL, USA, 35294. Phone : +1 205.934.2746 E-mail : [email protected] †The authors contributed equally to this work. bioRxiv preprint doi: https://doi.org/10.1101/2020.06.25.171942; this version posted June 27, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Inhibin Α Subunit Is Expressed by Bovine Ovarian Theca Cells and Its Knockdown Suppresses Androgen Production
www.nature.com/scientificreports OPEN ‘Free’ inhibin α subunit is expressed by bovine ovarian theca cells and its knockdown suppresses androgen production Mhairi Laird1, Claire Glister1, Warakorn Cheewasopit1,2, Leanne S. Satchell1, Andrew B. Bicknell1 & Phil G. Knight1* Inhibins are ovarian dimeric glycoprotein hormones that suppress pituitary FSH production. They are synthesised by follicular granulosa cells as α plus βA/βB subunits (encoded by INHA, INHBA, INHBB, respectively). Inhibin concentrations are high in follicular fuid (FF) which is also abundant in ‘free’ α subunit, presumed to be of granulosal origin, but its role(s) remains obscure. Here, we report the unexpected fnding that bovine theca cells show abundant INHA expression and ‘free’ inhibin α production. Thus, theca cells may contribute signifcantly to the inhibin α content of FF and peripheral blood. In vitro, knockdown of thecal INHA inhibited INSL3 and CYP17A1 expression and androgen production while INSL3 knockdown reduced INHA and inhibin α secretion. These fndings suggest a positive role of thecal inhibin α on androgen production. However, exogenous inhibin α did not raise androgen production. We hypothesised that inhibin α may modulate the opposing efects of BMP and inhibin on androgen production. However, this was not supported experimentally. Furthermore, neither circulating nor intrafollicular androgen concentrations difered between control and inhibin α-immunized heifers, casting further doubt on thecal inhibin α subunit having a signifcant role in modulating androgen production. Role(s), if any, played by thecal inhibin α remain elusive. Inhibins are glycoproteins of gonadal origin that play a key role in the negative feedback regulation of FSH pro- duction by pituitary gonadotrophs. -

The TGF-Β Family in the Reproductive Tract
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press The TGF-b Family in the Reproductive Tract Diana Monsivais,1,2 Martin M. Matzuk,1,2,3,4,5 and Stephanie A. Pangas1,2,3 1Department of Pathology and Immunology, Baylor College of Medicine, Houston, Texas 77030 2Center for Drug Discovery, Baylor College of Medicine, Houston, Texas 77030 3Department of Molecular and Cellular Biology, Baylor College of Medicine Houston, Texas 77030 4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030 5Department of Pharmacology, Baylor College of Medicine, Houston, Texas 77030 Correspondence: [email protected]; [email protected] The transforming growth factor b (TGF-b) family has a profound impact on the reproductive function of various organisms. In this review, we discuss how highly conserved members of the TGF-b family influence the reproductive function across several species. We briefly discuss how TGF-b-related proteins balance germ-cell proliferation and differentiation as well as dauer entry and exit in Caenorhabditis elegans. In Drosophila melanogaster, TGF-b- related proteins maintain germ stem-cell identity and eggshell patterning. We then provide an in-depth analysis of landmark studies performed using transgenic mouse models and discuss how these data have uncovered basic developmental aspects of male and female reproductive development. In particular, we discuss the roles of the various TGF-b family ligands and receptors in primordial germ-cell development, sexual differentiation, and gonadal cell development. We also discuss how mutant mouse studies showed the contri- bution of TGF-b family signaling to embryonic and postnatal testis and ovarian development. -

6 Signaling and BMP Antagonist Noggin in Prostate Cancer
[CANCER RESEARCH 64, 8276–8284, November 15, 2004] Bone Morphogenetic Protein (BMP)-6 Signaling and BMP Antagonist Noggin in Prostate Cancer Dominik R. Haudenschild, Sabrina M. Palmer, Timothy A. Moseley, Zongbing You, and A. Hari Reddi Center for Tissue Regeneration and Repair, Department of Orthopedic Surgery, School of Medicine, University of California, Davis, Sacramento, California ABSTRACT antagonists has recently been discovered. These are secreted proteins that bind to BMPs and reduce their bioavailability for interactions It has been proposed that the osteoblastic nature of prostate cancer with the BMP receptors. Extracellular BMP antagonists include nog- skeletal metastases is due in part to elevated activity of bone morphoge- gin, follistatin, sclerostatin, chordin, DCR, BMPMER, cerberus, netic proteins (BMPs). BMPs are osteoinductive morphogens, and ele- vated expression of BMP-6 correlates with skeletal metastases of prostate gremlin, DAN, and others (refs. 11–16; reviewed in ref. 17). There are cancer. In this study, we investigated the expression levels of BMPs and several type I and type II receptors that bind to BMPs with different their modulators in prostate, using microarray analysis of cell cultures affinities. BMP activity is also regulated at the cell membrane level by and gene expression. Addition of exogenous BMP-6 to DU-145 prostate receptor antagonists such as BAMBI (18), which acts as a kinase- cancer cell cultures inhibited their growth by up-regulation of several deficient receptor. Intracellularly, the regulation of BMP activity at cyclin-dependent kinase inhibitors such as p21/CIP, p18, and p19. Expres- the signal transduction level is even more complex. There are inhib- sion of noggin, a BMP antagonist, was significantly up-regulated by itory Smads (Smad-6 and Smad-7), as well as inhibitors of inhibitory BMP-6 by microarray analysis and was confirmed by quantitative reverse Smads (AMSH and Arkadia). -

Cumulin and FSH Cooperate to Regulate Inhibin B and Activin B Production by Human Granulosa-Lutein Cells in Vitro Dulama Richani
1 Cumulin and FSH cooperate to regulate inhibin B and activin B production by human 2 granulosa-lutein cells in vitro 3 4 Dulama Richani*1, Katherine Constance1, Shelly Lien1, David Agapiou1, William A. 5 Stocker2,3, Mark P. Hedger4, William L. Ledger1, Jeremy G. Thompson5, David M. 6 Robertson1, David G. Mottershead5,6, Kelly L. Walton2, Craig A. Harrison2 and Robert B. 7 Gilchrist1 8 9 1 Fertility and Research Centre, School of Women’s and Children’s Health, University of 10 New South Wales Sydney, Australia 11 2 Department of Physiology, Monash Biomedicine Discovery Institute, Monash University, 12 Clayton, Australia 13 3 Department of Chemistry and Biotechnology, Swinburne University of Technology, 14 Hawthorn, Victoria 3122, Australia 15 4 Centre for Reproductive Health, Hudson Institute of Medical Research, Clayton, VIC, 16 Australia 17 5 Robinson Research Institute, Adelaide Medical School, The University of Adelaide, 18 Australia 19 6 Institute for Science and Technology in Medicine, School of Pharmacy, Keele University, 20 Newcastle-under-Lyme, UK 21 22 Short Title: Cumulin and FSH jointly regulate inhibin/activin B 23 24 Corresponding author: 25 *Dulama Richani ([email protected]) 1 26 Wallace Wurth Building, UNSW Sydney, Kensington, NSW 2052, Australia 27 28 Re-print requests: 29 Dulama Richani ([email protected]) 30 Wallace Wurth Building, UNSW Sydney, Kensington, NSW 2052, Australia 31 32 Key words: 33 GDF9, BMP15, cumulin, FSH, inhibin, activin 34 35 Funding: 36 The work was funded by National Health and Medical Research Council of Australia grants 37 (APP1017484, APP1024358, APP1121504 awarded to DGM, CAH, and RBG, respectively) 38 and fellowships (APP1023210, APP1117538 awarded to RBG), and by Strategic Funds from 39 the University of New South Wales Sydney awarded to RBG. -

Inhibin Is a Novel Paracrine Factor for Tumor Angiogenesis and Metastasis
Author Manuscript Published OnlineFirst on March 13, 2018; DOI: 10.1158/0008-5472.CAN-17-2316 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Inhibin is a novel paracrine factor for tumor angiogenesis and metastasis Priyanka Singh1#, Laura M. Jenkins1#, Ben Horst1, Victoria Alers1, Shrikant Pradhan1, Prabhjot Kaur2, Tapasya Srivastava2, Nadine Hempel3, Balázs Győrffy4, Eugenia V. Broude5, Nam Y. Lee6, and Karthikeyan Mythreye5,1,* 1Department of Chemistry and Biochemistry, University of South Carolina, Columbia, South Carolina, USA 2Department of Genetics, University of Delhi, South Campus, India. 3Department of Pharmacology, Penn State University College of Medicine, Hershey PA 4MTA TTK Lendület Cancer Biomarker Research Group, Institute of Enzymology; Semmelweis University 2nd Department of Pediatrics, Budapest, Hungary. 5Department of Drug Discovery and Biomedical Sciences, School of Pharmacy, 6 Division of Pharmacology, College of Pharmacy, Ohio State University, Columbus, Ohio, USA # Equal contribution Running title: Inhibin promotes angiogenesis and metastasis * Correspondence to K. Mythreye: [email protected] Mailing address: Department of Chemistry and Biochemistry University of South Carolina, 631 Sumter Street Columbia SC 29208 Tel: (803) 576-5806, Fax: (803) 777-9521 Authors declare no conflict of interest. Keywords: Inhibin, Endoglin, ALK1, Angiogenesis, Ovarian cancer, Paracrine 1 Downloaded from cancerres.aacrjournals.org on September 24, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on March 13, 2018; DOI: 10.1158/0008-5472.CAN-17-2316 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract Inhibin is a heterodimeric TGF-β family ligand that is expressed in many cancers and is a selective biomarker for ovarian cancers, however its tumor-specific functions remain unknown. -

Context-Dependent Roles in Cell and Tissue Physiology
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press TGF-b and the TGF-b Family: Context-Dependent Roles in Cell and Tissue Physiology Masato Morikawa,1 Rik Derynck,2 and Kohei Miyazono3 1Ludwig Cancer Research, Science for Life Laboratory, Uppsala University, Biomedical Center, SE-751 24 Uppsala, Sweden 2Department of Cell and Tissue Biology, University of California at San Francisco, San Francisco, California 94143 3Department of Molecular Pathology, Graduate School of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan Correspondence: [email protected] The transforming growth factor-b (TGF-b) is the prototype of the TGF-b family of growth and differentiation factors, which is encoded by 33 genes in mammals and comprises homo- and heterodimers. This review introduces the reader to the TGF-b family with its complexity of names and biological activities. It also introduces TGF-b as the best-studied factor among the TGF-b family proteins, with its diversity of roles in the control of cell proliferation and differentiation, wound healing and immune system, and its key roles in pathology, for exam- ple, skeletal diseases, fibrosis, and cancer. lthough initially thought to stimulate cell TGF-b has been well documented in most cell Aproliferation, just like many growth factors, types, and has been best characterized in epithe- it became rapidly accepted that transforming lial cells. The bifunctional and context-depen- growth factor b (TGF-b) is a bifunctional reg- dent nature of TGF-b activities was further con- ulator that either inhibits or stimulates cell pro- firmed in a large variety of cell systems and liferation. -

Acvr2b (NM 007397) Mouse Tagged ORF Clone Lentiviral Particle Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for MR212153L3V Acvr2b (NM_007397) Mouse Tagged ORF Clone Lentiviral Particle Product data: Product Type: Lentiviral Particles Product Name: Acvr2b (NM_007397) Mouse Tagged ORF Clone Lentiviral Particle Symbol: Acvr2b Synonyms: 4930516B21Rik; ActRIIB Vector: pLenti-C-Myc-DDK-P2A-Puro (PS100092) ACCN: NM_007397 ORF Size: 1608 bp ORF Nucleotide The ORF insert of this clone is exactly the same as(MR212153). Sequence: OTI Disclaimer: The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. polymorphisms), each with its own valid existence. This clone is substantially in agreement with the reference, but a complete review of all prevailing variants is recommended prior to use. More info OTI Annotation: This clone was engineered to express the complete ORF with an expression tag. Expression varies depending on the nature of the gene. RefSeq: NM_007397.2 RefSeq Size: 1710 bp RefSeq ORF: 1611 bp Locus ID: 11481 UniProt ID: P27040 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Acvr2b (NM_007397) Mouse Tagged ORF Clone Lentiviral Particle – MR212153L3V Gene Summary: Transmembrane serine/threonine kinase activin type-2 receptor forming an activin receptor complex with activin type-1 serine/threonine kinase receptors (ACVR1, ACVR1B or ACVR1c). -

510(K) SUBSTANTIAL EQUIVALENCE DETERMINATION DECISION SUMMARY
510(k) SUBSTANTIAL EQUIVALENCE DETERMINATION DECISION SUMMARY A. 510(k) Number: K190661 B. Purpose for Submission: New Device C. Measurand: Somatic single nucleotide variants, insertions, deletions, and tumor mutational burden (TMB) in human genomic DNA obtained from formalin-fixed, paraffin-embedded tumor tissue. Refer to Appendix 1a for a complete list of the genes in the assay. D. Type of Test: Next generation sequencing tumor profiling test E. Applicant: NantHealth, Inc. F. Proprietary and Established Names: Trade Name: Omics Core Common Name: NantHealth Next Generating Sequencing Tumor Profiling Test G. Regulatory Information: 1. Regulation section: 21 CFR 866.6080 2. Classification: Class II 3. Product code: PZM 4. Panel: 88-Pathology 1 H. Intended Use: 1. Indications for use: The Omics Core assay is a qualitative in vitro diagnostic test that uses targeted next generation sequencing of formalin-fixed paraffin-embedded tumor tissue matched with normal specimens from patients with solid malignant neoplasms to detect tumor gene alterations in a broad multi gene panel. The test is intended to provide information on somatic mutations (point mutations and small insertions and deletions) and tumor mutational burden (TMB) for use by qualified health care professionals in accordance with professional guidelines, and is not conclusive or prescriptive for labeled use of any specific therapeutic product. Omics Core is a single-site assay performed at NantHealth, Inc. 2. Special conditions for use statement(s): For in vitro diagnostic use only For prescription use only 3. Special instrument requirements: Illumina NovaSeq6000 (qualified by NantHealth) I. Device Description: A description of required equipment, software, reagents, vendors, and storage conditions were provided, and are described in the product labeling (NantHealth Omics Core manual). -

Transforming Growth Factor- Β Type I Receptor Inhibitor Induces
대한남성과학회지:제 30 권 제 1 호 2012년 4월 Korean J Androl. Vol. 30, No. 1, April 2012 http://dx.doi.org/10.5534/kja.2012.30.1.23 Transforming Growth Factor-β Type I Receptor Inhibitor Induces Functional and Morphologic Recovery in a Rat Model of Erectile Dysfunction and Cavernous Fibrosis Ji Kan Ryu, Seung Min Oh, Hai Rong Jin, Kang Moon Song, Mi Hye Kwon, Do Kyung Kim, Jun Kyu Suh Department of Urology and National Research Center for Sexual Medicine, Inha University School of Medicine, Incheon, Korea =Abstract= Purpose: To examine the effectiveness of small-molecule inhibitor of transforming growth factor-β (TGF-β) type I receptor, an activin receptor-like kinase 5 (ALK5), on erectile dysfunction (ED) in a rat model of cavernous fibrosis, in which fibrosis was induced by intracavernous injection of adenovirus expressing TGF-β1 (Ad-TGF-β1). Materials and Methods: Four-month-old Sprague-Dawley rats were divided into four groups (n=10 per group): age-matched controls without treatment, age-matched controls receiving intracavernous injection of LacZ adenovirus, and cavernous fibrosis rats receiving an intracavernous injection of saline or ALK5 inhibitor (5 mg/kg). ALK5 inhibitor or saline was administered on day 5 after injection of Ad-TGF-β1. On day 30, erectile function was assessed by electrical stimulation of the cavernous nerve and the penis was then harvested for histologic studies (n=6 per group) and for the measurement of the hydroxyproline level (n=4 per group). Results: Ad-TGF-β1-induced cavernous fibrosis rats treated with saline showed a significant decrease in cavernous smooth muscle and endothelial content, and an increase in collagen deposition, which resulted in profound deterioration of all erectile function parameters, such as the ratios of maximal intracavernous pressure (ICP), total ICP, and slope to mean arterial pressure. -
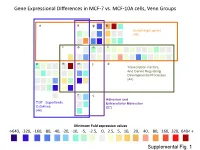
Supplementary Data
Gene Expressional Differences in MCF-7 vs. MCF-10A cells, Venn Groups e k g b Smad target genes (48) l o n i a h m j d Transcription Factors, And Genes Regulating Developmental Processes (44) f c Adhesion and TGF Superfamily Extracellular Molecules Cytokines (27) (44) Minimum Fold expression values ->640, -320, -160, -80, -40, -20, -10, -5, -2.5, 0, 2.5, 5, 10, 20, 40, 80, 160, 320, 640< + Supplemental Fig. 1 Supplemental Table 1. Change of Basal Gene Expressional values in MCF-7 as compared to MCF-10A cell line. Fold T-test Gene # GeneBank Symbol Up/Down p-Value Description /Position Regulation Venn Group a 01 /A01 NM_001105 ACVR1 -4.96 0.0000 Activin A receptor, type I 02 /A02 NM_001616 ACVR2A -2.62 0.0007 Activin A receptor, type IIA 03 /A03 NM_000020 ACVRL1 -1.25 0.7013 Activin A receptor type II-like 1 05 /A05 NM_020547 AMHR2 1.08 0.8043 Anti-Mullerian hormone receptor, type II 16 /B04 NM_004329 BMPR1A -2.40 0.0011 Bone morphogenetic protein receptor, type IA 17 /B05 NM_001203 BMPR1B -7.37 0.0000 Bone morphogenetic protein receptor, type IB 36 /C12 NM_000557 GDF5 -1.38 0.4911 Growth differentiation factor 5 (cartilage-derived morphogenetic protein-1) 37 /D01 NM_001001557 GDF6 1.12 0.9002 Growth differentiation factor 6 38 /D02 NM_182828 GDF7 -1.52 0.4995 Growth differentiation factor 7 53 /E05 NM_020997 LEFTY1 -1.76 0.0529 Left-right determination factor 1 59 /E11 NM_018055 NODAL -3.62 0.1290 Nodal homolog (mouse) 77 /G05 NM_003238 TGFB2 -4.52 0.0566 Transforming growth factor, beta 2 78 /G06 NM_003239 TGFB3 -1.12 0.2902 Transforming