Synthesis of Glycosides of Glucuronic, Galacturonic and Mannuronic Acids: an Overview
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GRAS Notice 789 for Erythritol
GRAS Notice (GRN) No. 789 https://www.fda.gov/food/generally-recognized-safe-gras/gras-notice-inventory. Toi• Strategies ~~~~G~~[)) JUN 7 20'8 Innovative solutions Sound science OFFICE OF FOOD ADDITIVE SAFE1Y June 5, 2018 Dr. Dennis Keefe Director, Division of Biotechnology and GRAS Notice Review Office of Food Additive Safety (HFS-200) Center for Food Safety and Applied Nutrition Food and Drug Administration 5100 Paint Branch Parkway College Park, MD 20740-3835 Subject: GRAS Notification - Erythritol Dear Dr. Keefe: On behalf of Cargill, Incorporated, ToxStrategies, Inc. (its agent) is submitting, for FDA review, a copy of the GRAS notification as required. The enclosed document provides notice of a claim that the food ingredient, erythritol, described in the enclosed notification is exempt from the premarket approval requirement of the Federal Food, Drug, and Cosmetic Act because it has been determined to be generally recognized as safe (GRAS), based on scientific procedures, for addition to food. If you have any questions or require additional information, please do not hesitate to contact me at 630-352-0303, or [email protected]. Sincerely, (b) (6) Donald F. Schmitt, M.P.H. Senior Managing Scientist ToxStrategies, Inc., 931 W. 75th St. , Suite 137, PMB 263, Naperville, IL 60565 1 Office (630) 352-0303 • www.toxstrategies.com GRAS Determination of Erythritol for Use in Human Food JUNES,2018 Innovative solutions s ,..,.,',--.r-.r--.r--. OFFICE OF FOOD ADDITIVE SAFE1Y GRAS Determination of Erythritol for Use in Human Food SUBMITTED BY: Cargill, Incorporated 15407 McGinty Road West Wayzata, MN 55391 SUBMITTED TO: U.S. Food and Drug Administration Center for Food Safety and Applied Nutrition Office of Food Additive Safety HFS-200 5100 Paint Branch Parkway College Park MD 20740-3835 CONTACT FOR TECHNICAL OR OTIIER INFORMATION Donald F. -

Phthalates: Toxicology and Food Safety – a Review
Czech J. Food Sci. Vol. 23, No. 6: 217–223 Phthalates: Toxicology and Food Safety – a Review PŘEMYSL MIKULA1, ZDEŇKA SVOBODOVÁ1 and MIRIAM SMUTNÁ2 1Department of Veterinary Public Health and Toxicology and 2Department of Biochemistry and Biophysics, Faculty of Veterinary Hygiene and Ecology, University of Veterinary and Pharmaceutical Sciences, Brno, Czech Republic Abstract MIKULA P., SVOBODOVÁ Z., SMUTNÁ M. (2005): Phthalates: toxicology and food safety – a review. Czech J. Food Sci., 23: 217–223. Phthalates are organic substances used mainly as plasticisers in the manufacture of plastics. They are ubiquitous in the environment. Although tests in rodents have demonstrated numerous negative effects of phthalates, it is still unclear whether the exposure to phthalates may also damage human health. This paper describes phthalate toxicity and toxicokinetics, explains the mechanisms of phthalate action, and outlines the issues relating to the presence of phthalates in foods. Keywords: di(2-ethylhexyl)phthalate; dibutyl phthalate; peroxisome proliferators; reproductive toxicity Phthalic acid esters, often also called phtha- air, soil, food, etc.). People as well as animals lates, are organic substances frequently used can be exposed to these compounds through in many industries. They are usually colourless ingestion, inhalation or dermal exposure, and or slightly yellowish oily and odourless liquids iatrogenic exposure to phthalates from blood only very slightly soluble in water. Phthalates are bags, injection syringes, intravenous canyllas much more readily soluble in organic solvents, and catheters, and from plastic parts of dialysers and the longer their side chain, the higher their is also a possibility (VELÍŠEK 1999; ČERNÁ 2000; liposolubility and the boiling point. Phthalates LOVEKAMP-SWAN & DAVIS 2003). -

The Disposition of Morphine and Its 3- and 6-Glucuronide Metabolites in Humans
>+'è ,'i5 1. The Disposition of Morphine and its 3- and 6- Glucuronide Metabolites in Humans and Sheep A Thesis submitted in Fulfilment of the Requirements for the Degree of Doctor of Philosophy in Science at the University of Adelaide Robert W. Milne B.Pharm.(Hons), M.Sc. Department of Clinical and Experimental Pharmacology University of Adelaide August 1994 Ar,-,..u i.'..t itttlí I Corrigenda Page 4 The paragraph at the top of the page should be relocated to inimediatèly follow the section heading "1.2 Chemistry" on page 3" Page 42 (lines 2,5 and/) "a1-acid glycoProtein" should read "cr1-acid glycoprotein". Page 90 In the footnote "Amoxicillin" and "Haemacell" should read "Amoxycillin" and "Haemaccel", respectively. Page I 13 (tine -l l) "supernate!' should read "supernatant". ' '(j- Page I 15 (line -9) "supernate" should read "supernatalït1-. Page 135 To the unfinished sentence at the bottom of the page should be added "appears to be responsible for the formation of M3G, but its relative importance remains unknown. It was also proposed that morphine is involved in an enterohepatic cycle." Page 144 Add "Both infusions ceased at 6 hr" to the legend to Figure 6.3. Page 195 (line 2) "was" should fead "were". Page 198 (line l3) "ro re-€xamine" should read "to te-examine". Pages 245 to29l Bibliosraohv: "8r.." should read "Br.". For the reference by Bhargava et aI. (1991), "J. Pharmacol.Exp.Ther." should read "J. Pharmacol. Exp. Ther." For the reference by Chapman et aI. (1984), a full-stop should appear after"Biomed" . For the reference by Murphey et aI. -
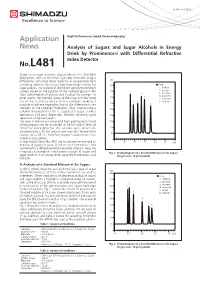
Analysis of Sugars and Sugar Alcohols in Energy Drink by Prominence-I with Differential Refreactive Index Detector
LAAN-A-LC-E258 Application High Performance Liquid Chromatography News Analysis of Sugars and Sugar Alcohols in Energy Drink by Prominence-i with Differential Refractive Index Detector No.L481 Sugars and sugar alcohols display almost no ultraviolet absorption, and are therefore typically detected using a differential refractive index detector or evaporative light uRI scattering detector. By using a ligand exchange column for 80 ■ Peaks sugar analysis, it is possible to distinguish among the different 1. maltose 70 2. glucose isomers based on the position of the hydroxyl group in the 1 4 3. fructose chair conformation of glucose and fructose for example. In 4. erythritol 60 5 other words, the hydroxyl group of the sugar and the metal 5. mannitol ion of the stationary phase form a complex, making it 2 3 6. sorbitol possible to achieve separation due to the difference in the 50 6 strength of the complex formation. Also, maintaining a 40 column temperature of 80 °C suppresses sugar anomer separation and peak dispersion, thereby achieving good 30 separation of adjacent peaks. The new Prominence-i integrated high-performance liquid 20 chromatograph can be connected to the RID-20A differential refractive index detector. The column oven, which can 10 accommodate a 30 cm column and maintain temperature 0 control up to 85 °C, therefore supports applications that require a long column. In Application News No. 467, we introduced an example of 0 5 10 15 20 25 min analysis of sugars in juice, in which the Prominence-i was connected to a differential refractive index detector. Here, we introduce an example of simultaneous analysis of sugars and Fig. -

United States Patent Office
United States Patentaw Office ... ber of ether groupings on each molecule. These mix tures, however, if they analyze as containing an average 2,978,421. number of ether groups per molecule greater than one, NITRILE COPOLYMERS AND METHOD OF are capable of producing the insoluble nitrile polymers PREPARING SAME of this invention. Since the efficiency of the polyether cross-linking agent increases with the number of poten John A. Holloway, Cleveland, Ohio, assignor to The tially polymerizable groups on the molecule, it is much B. F. Goodrich Company, New York, N.Y., a corpo preferred to utilize polyethers containing an average of ration of New York - two or more alkenyl ether groupings per molecule. The No Drawing. Filed June 9, 1958, Ser. No. 740,566. 10 polyvinyl polyethers of the polyhydric alcohols within the above broad class are produced by reacting acetylene 9 Claims. (C. 260-17.4) with the polyhydric alcohol (or an alcoholate thereof) in a Reppe-type vinylation synthesis. The polycrotyl ethers of the polyhydric alcohols are also useful although they This invention relates to cross-linked alpha-beta ole 15 do not contain a terminal CHCC group. - finically unsaturated nitrile polymers and more particu Illustrative polyhydric alcohols of the above-described larly pertains to copolymers of alpha-beta olefinically class that may be utilized in the preparation of the poly. unsaturated nitriles and polyalkenyl polyethers of poly alkenyl polyether, cross-linking agent include the butane hydric alcohols and methods for their preparation. triols such as 1,2,3-butane triol, 2,3,4-trihydroxybutyric An object of this invention is the provision of insoluble, 20 acid, the aldotetroses such as erythrose and threose, keto cross-linked polynitriles which are capable of thickening tetroses such as erythrulose; the aldopentoses such as certain non-aqueous polar solvents. -

Safety Evaluation of Certain Food Additives and Contaminants. WHO Food Additives Series, No
WHO FOOD Safety evaluation of ADDITIVES certain food additives SERIES: 64 and contaminants Prepared by the Seventy-third meeting of the Joint FAO/ WHO Expert Committee on Food Additives (JECFA) World Health Organization, Geneva, 2011 WHO Library Cataloguing-in-Publication Data Safety evaluation of certain food additives and contaminants / prepared by the seventy-third meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). (WHO food additives series ; 64) 1.Food additives - toxicity. 2.Food contamination. 3.Flavoring agents - analysis. 4.Flavoring agents - toxicity. 5.Risk assessment. I.Joint FAO/WHO Expert Committee on Food Additives. Meeting (73rd : 2010: Geneva, Switzerland). II.World Health Organization. III.Series. ISBN 978 924 166064 8 (NLM classification: WA 712) ISSN 0300-0923 © World Health Organization 2011 All rights reserved. Publications of the World Health Organization can be obtained from WHO Press, World Health Organization, 20 Avenue Appia, 1211 Geneva 27, Switzerland (tel.: +41 22 791 3264; fax: +41 22 791 4857; e-mail: [email protected]). Requests for permission to reproduce or translate WHO publications—whether for sale or for non- commercial distribution—should be addressed to WHO Press at the above address (fax: +41 22 791 4806; e-mail: [email protected]). The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. -

LIQUID-VAPOR EQUILIBRIUM THERMODYNAMICS of the FUSEL OIL: a CASE STUDY Jéssyka Jennifer Miranda Corrêa¹, Edilailsa Januário
LIQUID-VAPOR EQUILIBRIUM THERMODYNAMICS OF THE FUSEL OIL: A CASE STUDY Jéssyka Jennifer Miranda Corrêa¹, Edilailsa Januário de Melo², José Izaquiel Santos da Silva³ ¹Graduating in Chemical Engineering from the Federal University of the Jequitinhonha and Mucuri Valleys, Diamantina - MG, Brazil. ²PhD student in Chemical Engineering, College of Chemical Engineering - State University of Campinas - SP, Brazill. ³Professor at the Federal University of the Jequitinhonha and Mucuri Valleys, Diamantina - MG, Brazil. E-mail for contact: [email protected] Recebido em: 06/04/2019 – Aprovado em: 10/06/2019 – Publicado em: 30/06/2019 DOI: 10.18677/EnciBio_2019A183 ABSTRACT Fusel oil is a by-product of the process of recovering hydrated ethanol, consisting of a mixture of superior alcohols, ethanol, water, among other components. Its commercial interest is mainly due to the presence of isoamyl alcohol, one of the raw materials used in the synthesis of esters, which compounds are relevant for the chemical industry. The recovery of this superior alcohol involves unit operations based on phase equilibrium, which for multicomponent systems can be studied in software that simulates the operation of equipment, in order to present predictions of interactions between its components, reducing errors, time and costs of design. In the industrial, academic and scientific fields, there are numerous challenges regarding the design of separation processes and the monitoring of mixing effects in typical operations, and the Aspen Plus process simulation and optimization platform excels in solving these issues. The current study aims to predict the Liquid-Vapor Equilibrium of the fusel oil, which was considered as a mixture of five components, from the behavioral analysis of each of the binaries that compose it. -

Glucuronic Acid Derivatives in Enzymatic Biomass Degradation: Synthesis and Evaluation of Enzymatic Activity
Downloaded from orbit.dtu.dk on: Oct 08, 2021 Glucuronic Acid Derivatives in Enzymatic Biomass Degradation: Synthesis and Evaluation of Enzymatic Activity d'Errico, Clotilde Publication date: 2016 Document Version Publisher's PDF, also known as Version of record Link back to DTU Orbit Citation (APA): d'Errico, C. (2016). Glucuronic Acid Derivatives in Enzymatic Biomass Degradation: Synthesis and Evaluation of Enzymatic Activity. DTU Chemistry. General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. Users may download and print one copy of any publication from the public portal for the purpose of private study or research. You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. GLUCURONIC ACID DERIVATIVES IN ENZYMATIC BIOMASS DEGRADATION: SYNTHESIS AND EVALUATION OF ENZYMATIC ACTIVITY PHD THESIS – MAY 2016 CLOTILDE D’ERRICO DEPARTMENT OF CHEMISTRY TECHNICAL UNIVERSITY OF DENMARK ACKNOWLEDGEMENTS This dissertation describes the work produced during my PhD studies. The work was primarily conducted at the DTU Chemistry in the period between November 2012 and January 2016 with an external stay at Novozymes A/S between September 2013 and March 2014. -

IPOLYHYDRIC ALCOHOLS from WOOD July 1954
IPOLYHYDRIC ALCOHOLS FROM WOOD July 1954 DEC 221954 No. 1984 UNITED STATES DEPARTMENT OF AGRICULTURE --,LFOREST. SERVICE 40REST PRODUCTS LABORATORY÷Q-.---,Y) Madison 5,Wisconsin In Cooperation with the University of Wisconsin 1 POLYHYDRIC ALCOHOLS FROM WOOD-2 2— By J. A. HALL, Director Forest Products Laboratory, 3 Forest Service U. S. Department of Agriculture Wood-sugar solutions produced in the laboratory-scale pilot plant at the U. S. Forest Products Laboratory have been proven to be satis- factory for the production of industrial alcohol, feeding yeast, and wood 4 molasses for stock feed (14, 15, 16, P 17). ± As a continuation of the pro- gram of the Laboratory for developing chemical utilization processes for the wood residues resulting from normal logging operations, and lumber, veneer, and the other forest products manufacture, investiga- tions are in progress to convert wood sugar into merchantable products. Because of the versatility and greatly increased demand for polytlydric alcohols, a study of the production of these materials has been chosen. The alkyd resin industry in the United States has increased tenfold in the past few years and no evidence of decline has been noted. Recently sorbitol has been made available commercially by the catalytic hydro- genation of corn sugar (2). The annual capacity now is 75 million pounds. Sorbitol is widely used as a humectant and conditioner in tobacco, cello- phane, and other products (3). However, one of its fast growing uses is for alkyd resins, where it may replace up to 50 percent glycerine and 25 percent pentaerythritol. This is fortunate since the production of glycerine in the United States as a byproduct of the soap manufacturing 1Presented before the Food and Agriculture Organization of the United Nations, Expert Panel on the Chemistry of Wood, meeting in Stockholm, Sweden, July 27-28, 1953. -

Hydrolytic Enzymes Targeting to Prodrug/Drug Metabolism for Translational Application in Cancer
Journal of Clinical Science & Translational Medicine Hydrolytic Enzymes Targeting to Prodrug/Drug Metabolism for Translational Application in Cancer Prabha M* Review Article Department of Biotechnology, Ramaiah Institute of Technology, India Volume 1 Issue 1 Received Date: April 04, 2019 *Corresponding author: Prabha M, Department of Biotechnology, Ramaiah Institute of Published Date: June 20, 2019 Technology, Bangalore-560054, India, Tel: 080-23588236; Email: [email protected] Abstract A variety of approaches are under development to improve the effectiveness and specificity of enzyme activation for drug metabolism on tumour cell targeting for cancer treatment. Many such methods involve conjugates like monoclonal antibodies, substrate specificity and offer attractive means of directing to tumour toxic agents such as drugs, radioisotopes, protein cytotoxins, cytokines, effector cells of the immune system, gene therapy, stem cell therapy and enzyme therapy for therapeutic use. Hydrolytic enzymes belong to class III of enzyme classification and play an important role in the drug metabolism towards the treatment of cancer. Hydrolases helps drugs for metabolic efficiency to target on cancer cell since it is involved in the hydrolytic reaction of various biomolecules and compounds. The prodrug is designed to be a substrate for the chosen enzyme activity. A number of prodrugs have been developed that can be transformed into an active anticancerous drugs by enzymes of both mammalian and non mammalian origin. The basic molecular biochemistry, biotechnological processes and other information related to enzyme catalysis, has a major impact for the production of efficient drugs. In the current review some of the Hydrolases has been discussed which play a significant role towards prodrug to drug metabolism for cancer treatment. -

In Vitroo and in Vivo Pharmacology of 4-Substituted
IN VITROO AND IN VIVO PHARMACOLOGY OF 4-SUBSTITUTED METHOXYBENZOYL-ARYL-THIAZOLES (SMART) AND 2- ARYLTHIAZOLIDINE-4-CARBOXYLIC ACID AMIDES (ATCAA) Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Chien-Ming Li, M.S. Graduate Program in Pharmacy The Ohio State University 2010 Dissertation Committee: Dr. James T. Dalton, Advisor Dr. Robert W. Brueggemeier Dr. Thomas D. Schmittgen Dr. Mitch A. Phelps Copyright by Chien-Ming Li 2010 ABSTRACT Formation of microtubules is a dynamic process that involves polymerization and depolymerization of the αβ-tubulin heterodimers. Drugs that enhance or inhibit tubulin polymerization can destroy this dynamic process, arresting cells in the G2/M phase of the cell cycle. Although drugs that target tubulin generally demonstrate cytotoxic potency in sub-nanomolar range, resistance due to drug efflux is a common phenomenon among the antitubulin agents. We recently reported a class of 4-Substituted Methoxybenzoyl-Aryl- Thiazoles (SMART), which exhibited great in vitro and in vivo potency. SMART compounds effectively inhibited tubulin polymerization in dose dependent manner, suggesting that SMART compounds may bind to tubulin and subsequently hamper the polymerization. To date the only method to determine the binding of inhibitor to tubulin has been competitive radioligand binding assays. We developed a novel non-radioactive mass spectrometry (MS) binding assay to study the tubulin binding of colchicine, vinblastine and paclitaxel. The method involves a very simple step of separating the unbound ligand from macromolecules using ultrafiltration. The unbound ligand in the filtrate can be accurately determined using a highly sensitive and specific LC-MS/MS method. -

Properties and Kinetic Analysis of UDP-Glucose Dehydrogenase from Group a Streptococci IRREVERSIBLE INHIBITION by UDP-CHLOROACETOL*
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 272, No. 6, Issue of February 7, pp. 3416–3422, 1997 © 1997 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. Properties and Kinetic Analysis of UDP-glucose Dehydrogenase from Group A Streptococci IRREVERSIBLE INHIBITION BY UDP-CHLOROACETOL* (Received for publication, September 19, 1996, and in revised form, November 6, 1996) Robert E. Campbell‡§, Rafael F. Sala‡, Ivo van de Rijn¶, and Martin E. Tanner‡i From the ‡Department of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z1, Canada and ¶Wake Forest University Medical Center, Winston-Salem, North Carolina 27157 UDP-glucuronic acid is used by many pathogenic bac- the capsule enables the bacteria to evade the host’s immune teria in the construction of an antiphagocytic capsule system (7, 8). Group A and C streptococci are mammalian that is required for virulence. The enzyme UDP-glucose pathogens that use UDPGDH in the synthesis of a capsule dehydrogenase catalyzes the NAD1-dependent 2-fold ox- composed of hyaluronic acid (a polysaccharide consisting of idation of UDP-glucose and provides a source of the alternating glucuronic acid and N-acetylglucosamine residues) acid. In the present study the recombinant dehydrogen- (9, 10). Many of the known strains of Streptococcus pneumoniae ase from group A streptococci has been purified and also use UDP-glucuronic acid in the construction of their po- found to be active as a monomer. The enzyme contains lysaccharide capsule (11), and it has recently been shown that no chromophoric cofactors, and its activity is unaffected UDPGDH is required for capsule production in S.