Elevated Expression of Toxin Tisb Protects Persister Cells Against Ciprofloxacin but Enhances Susceptibility to Mitomycin C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Use of Ceragenins As a Potential Treatment for Urinary Tract Infections Urszula Wnorowska1, Ewelina Piktel1, Bonita Durnaś2, Krzysztof Fiedoruk3, Paul B
Wnorowska et al. BMC Infectious Diseases (2019) 19:369 https://doi.org/10.1186/s12879-019-3994-3 RESEARCH ARTICLE Open Access Use of ceragenins as a potential treatment for urinary tract infections Urszula Wnorowska1, Ewelina Piktel1, Bonita Durnaś2, Krzysztof Fiedoruk3, Paul B. Savage4 and Robert Bucki1* Abstract Background: Urinary tract infections (UTIs) are one of the most common bacterial infections. High recurrence rates and the increasing antibiotic resistance among uropathogens constitute a large social and economic problem in current public health. We assumed that combination of treatment that includes the administration ceragenins (CSAs), will reinforce the effect of antimicrobial LL-37 peptide continuously produced by urinary tract epithelial cells. Such treatment might be an innovative approach to enhance innate antibacterial activity against multidrug- resistant E. coli. Methods: Antibacterial activity measured using killing assays. Biofilm formation was assessed using crystal violet staining. Viability of bacteria and bladder epithelial cells subjected to incubation with tested agents was determined using MTT assays. We investigated the effects of chosen molecules, both alone and in combinations against four clinical strains of E. coli, obtained from patients diagnosed with recurrent UTI. Results: We observed that the LL-37 peptide, whose concentration increases at sites of urinary infection, exerts increased bactericidal effect against E. coli when combined with ceragenins CSA-13 and CSA-131. Conclusion: We suggest that the employment of combination of natural peptide LL-37 with synthetic analogs might be a potential solution to treat urinary tract infections caused by drug-resistant bacteria. Keywords: Urinary tract infection, LL-37 peptide, Ceragenins, Bacterial drug resistance Background trimethoprim/sulphamethoxazole, may no longer be used Urinary tract infections (UTIs) are one of the most com- for empiric treatment due to high resistance rates [7]. -

Intravenous Palonosetron Increases the Incidence of Qtc Prolongation During Sevoflurane General Anesthesia for Laparotomy
Korean J Anesthesiol 2013 November 65(5): 397-402 Clinical Research Article http://dx.doi.org/10.4097/kjae.2013.65.5.397 Intravenous palonosetron increases the incidence of QTc prolongation during sevoflurane general anesthesia for laparotomy Jeong Jin Min, Yongjae Yoo, Tae Kyong Kim, and Jung-Man Lee Department of Anesthesiology and Pain Medicine, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea Background: Palonosetron is a recently introduced 5-hydroxytryptamine-3 (5-HT3) receptor antagonist useful for postoperative nausea and vomiting prophylaxis. However, 5-HT3 receptor antagonists increase the corrected QT (QTc) interval in patients who undergo general anesthesia. This retrospective study was performed to evaluate whether palono- setron would induce a QTc prolongation in patients undergoing general anesthesia with sevoflurane. Methods: We reviewed a database of 81 patients who underwent general anesthesia with sevoflurane. We divided the records into palonosetron (n = 41) and control (n = 40) groups according to the use of intraoperative palonosetron, and analyzed the electrocardiographic data before anesthesia and 30, 60, 90, and 120 min after skin incision. Changes in the QTc interval from baseline, mean blood pressure, heart rate, body temperature, and sevoflurane concentrations at each time point were compared between the two groups. Results: The QTc intervals at skin incision, and 30, 60, 90, and 120 min after the skin incision during general anesthesia were significantly longer than those at baseline in the two groups (P < 0.001). The changes in the QTc intervals were not different between the two groups (P = 0.41). However, six patients in the palonosetron group showed a QTc interval > 500 ms 30 min after skin incision, whereas no patient did in the control group (P = 0.01). -

Antibacterial Effect of Sevoflurane and Isoflurane Ángel Martínez-Monsalve3 María Dolores Crespo- Sánchez1
Original María Martínez-Serrano1 Manuel Gerónimo-Pardo2 Antibacterial effect of sevoflurane and isoflurane Ángel Martínez-Monsalve3 María Dolores Crespo- Sánchez1 1Servicio de Microbiología y Parasitología. Complejo Hospitalario Universitario de Albacete. 2Servicio de Anestesiología y Reanimación. Complejo Hospitalario Universitario de Albacete. 3Servicio de Cirugía Vascular. Complejo Hospitalario Universitario de Albacete. ABSTRACT property in vivo. This might then allow these agents to be con- sidered as rescue treatment against multidrug resistant patho- Introduction. Multidrug resistant bacteria are increasing gens, including a topical use in infected wounds. worldwide and therapeutic options are limited. Some anaes- Key words: Sevoflurane, Isoflurane, Anaesthetics, Inhala- thetics have shown antibacterial activity before. In this study, tion, Anti-Infective Agents. we have investigated the antibacterial effect of the halogen- ated anaesthetic agents sevoflurane and isoflurane against a range of resistant pathogens. Actividad antibacteriana de sevoflurano e Methods. Two experiments were conducted. In the first, isoflurano bacterial suspensions of both ATCC and resistant strains of Staphylococcus aureus, Escherichia coli and Pseudomonas RESUMEN aeruginosa were exposed to liquid sevoflurane and isoflurane during 15, 30 and 60 minutes. In the second experiment clinical Introducción. Las bacterias multirresistentes están au- resistant strains of E. coli, Klebsiella pneumoniae, Enterobac- mentando en todo el mundo y las opciones terapéuticas son ter cloacae, P. aeruginosa, Acinetobacter baumannii, S. aureus, limitadas. Algunos anestésicos han mostrado actividad an- and Enterococcus faecium were studied. Previously inoculated tibacteriana previamente. En este estudio hemos investigado agar plates were irrigated with the halogenated anaesthet- dicha actividad en los anestésicos halogenados sevoflurano e ic agents and these were left to evaporate before the plates isoflurano frente a un grupo de patógenos resistentes. -

Antibacterial Activity and Mechanisms of Essential Oil from Citrus Medica L
molecules Article Antibacterial Activity and Mechanisms of Essential Oil from Citrus medica L. var. sarcodactylis Ze-Hua Li 1,2, Ming Cai 2,*, Yuan-Shuai Liu 3, Pei-Long Sun 2,* and Shao-Lei Luo 2 1 College of Biotechnology and Bioengineering, Zhejiang University of Technology, Hangzhou 310014, China; [email protected] 2 Department of Food Science and Technology, Zhejiang University of Technology, Hangzhou 310014, China; [email protected] 3 Department of Chemical and Biological Engineering, Hong Kong University of Science and Technology, Hong Kong; [email protected] * Correspondence: [email protected] (P.-L.S.); [email protected] (M.C.); Tel.: +86-0571-88320388 (P.-L.S.); +86-0571-88320345 (M.C.) Academic Editor: Francesca Mancianti Received: 29 March 2019; Accepted: 18 April 2019; Published: 22 April 2019 Abstract: In this work, antibacterial activity of finger citron essential oil (FCEO, Citrus medica L. var. sarcodactylis) and its mechanism against food-borne bacteria were evaluated. A total of 28 components in the oil were identified by gas chromatography-mass spectrometry, in which limonene (45.36%), γ-terpinene (21.23%), and dodecanoic acid (7.52%) were three main components. For in vitro antibacterial tests, FCEO exhibited moderately antibacterial activity against common food-borne bacteria: Escherichia coli, Staphylococcus aureus, Bacillus subtilis and Micrococcus luteus. It showed a better bactericidal effect on Gram-positive bacteria than Gram-negative. Mechanisms of the antibacterial action were investigated by observing changes of bacteria morphology according to scanning electron microscopy, time-kill analysis, and permeability of cell and membrane integrity. Morphology of tested bacteria was changed and damaged more seriously with increased concentration and exposure time of FCEO. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0296.191 A1 PATEL Et Al
US 20140296.191A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0296.191 A1 PATEL et al. (43) Pub. Date: Oct. 2, 2014 (54) COMPOSITIONS OF PHARMACEUTICAL (52) U.S. Cl. ACTIVES CONTAINING DETHYLENE CPC ............... A61K 47/10 (2013.01); A61 K9/0019 GLYCOL MONOETHYLETHER OR OTHER (2013.01); A61 K9/0048 (2013.01); A61 K ALKYL DERVATIVES 45/06 (2013.01) USPC ........... 514/167: 514/177; 514/178: 514/450; (71) Applicant: THEMIS MEDICARE LIMITED, 514/334: 514/226.5: 514/449; 514/338; Mumbai (IN) 514/256; 514/570; 514/179; 514/174: 514/533; (72) Inventors: Dinesh Shantilal PATEL, Mumbai (IN); 514/629; 514/619 Sachin Dinesh PATEL, Mumbai (IN); Shashikant Prabhudas KURANI, Mumbai (IN); Madhavlal Govindlal (57) ABSTRACT PATEL, Mumbai (IN) (73) Assignee: THEMIS MEDICARE LIMITED, The present invention relates to pharmaceutical compositions Mumbai (IN) of various pharmaceutical actives, especially lyophilic and hydrophilic actives containing Diethylene glycol monoethyl (21) Appl. No.: 14/242,973 ether or other alkyl derivatives thereofas a primary vehicle and/or to pharmaceutical compositions utilizing Diethylene (22) Filed: Apr. 2, 2014 glycol monoethyl ether or other alkyl derivatives thereofas a primary vehicle or as a solvent system in preparation of Such (30) Foreign Application Priority Data pharmaceutical compositions. The pharmaceutical composi Apr. 2, 2013 (IN) ......................... 1287/MUMA2013 tions of the present invention are safe, non-toxic, exhibits enhanced physical stability compared to conventional formu Publication Classification lations containing such pharmaceutical actives and are Suit able for use as injectables for intravenous and intramuscular (51) Int. Cl. administration, as well as for use as a preformed solution/ A647/ (2006.01) liquid for filling in and preparation of capsules, tablets, nasal A6 IK 45/06 (2006.01) sprays, gargles, dermal applications, gels, topicals, liquid oral A6 IK9/00 (2006.01) dosage forms and other dosage forms. -

Antimicrobials: Killing Persisters While They Sleep
RESEARCH HIGHLIGHTS ANTIMICROBIALS Killing persisters while they sleep Bacterial persisters are a certain metabolites can enhance of mannitol enhanced gentamicin subpopulation of dormant cells the killing of both Gram-negative killing of persister cells by more than that have been implicated in a and Gram-positive persisters by two orders of magnitude. Similarly, it may be possible range of chronic and recurrent aminoglycosides. in mice that were implanted with to eradicate infections through their ability Aminoglycoside uptake into the catheters which had been colonized bacterial to survive antibiotic treatments. bacterium is energy dependent, by a uropathogenic E. coli strain, Although most cellular processes are leading the authors to investigate administration of mannitol together persisters in a completely shut down in persisters, whether metabolic stimulation with gentamicin reduced the viability clinical setting translation still occurs, albeit at a enhances the killing of persister of biofilm bacteria on the catheter by stimulating reduced rate, making the use of cells by increasing the uptake of by more than an order of magnitude. their underlying aminoglycoside antibiotics (which these antibiotics. They found that Finally, the authors tested whether metabolic activity target the ribosome) an attractive the addition of glucose, mannitol, metabolite addition enhanced option. However, aminoglycosides fructose or pyruvate increased the aminoglycoside killing of Gram- concurrently have only weak activity against this killing of isolated Escherichia coli positive bacteria; whereas mannitol, with antibiotic subpopulation of cells. Writing persisters by gentamicin, kanamycin glucose and pyruvate had no effect, treatment. in Nature, Collins and colleagues and streptomycin by more than three the addition of fructose enhanced now show that the addition of orders of magnitude. -

Jp Xvii the Japanese Pharmacopoeia
JP XVII THE JAPANESE PHARMACOPOEIA SEVENTEENTH EDITION Official from April 1, 2016 English Version THE MINISTRY OF HEALTH, LABOUR AND WELFARE Notice: This English Version of the Japanese Pharmacopoeia is published for the convenience of users unfamiliar with the Japanese language. When and if any discrepancy arises between the Japanese original and its English translation, the former is authentic. The Ministry of Health, Labour and Welfare Ministerial Notification No. 64 Pursuant to Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (Law No. 145, 1960), the Japanese Pharmacopoeia (Ministerial Notification No. 65, 2011), which has been established as follows*, shall be applied on April 1, 2016. However, in the case of drugs which are listed in the Pharmacopoeia (hereinafter referred to as ``previ- ous Pharmacopoeia'') [limited to those listed in the Japanese Pharmacopoeia whose standards are changed in accordance with this notification (hereinafter referred to as ``new Pharmacopoeia'')] and have been approved as of April 1, 2016 as prescribed under Paragraph 1, Article 14 of the same law [including drugs the Minister of Health, Labour and Welfare specifies (the Ministry of Health and Welfare Ministerial Notification No. 104, 1994) as of March 31, 2016 as those exempted from marketing approval pursuant to Paragraph 1, Article 14 of the Same Law (hereinafter referred to as ``drugs exempted from approval'')], the Name and Standards established in the previous Pharmacopoeia (limited to part of the Name and Standards for the drugs concerned) may be accepted to conform to the Name and Standards established in the new Pharmacopoeia before and on September 30, 2017. -

Mechanisms of Staphylococcus Aureus Persistence And
MECHANISMS OF STAPHYLOCOCCUS AUREUS PERSISTENCE AND ERADICATION OF CHRONIC STAPHYLOCOCCAL INFECTIONS by Rebecca Yee A dissertation submitted to the Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland December, 2018 ABSTRACT Bacteria can exist in different phenotypic states depending on environmental conditions. Under stressed conditions, such as antibiotic exposure, bacteria can develop into persister cells that allow them to stay dormant until the stress is removed, when they can revert back to a growing state. The interconversion of non-growing persister cells and actively growing cells is the underlying basis of relapsing and chronic persistent infections. Eradication and better treatment of chronic, persistent infections caused by Staphylococcus aureus requires a multi- faceted approach, including a deeper understanding of how the bacteria persist under stressed conditions, regulate its cell death pathways, and development of novel drug therapies. Persisters were first discovered in the 1940s in a staphylococcal culture in which penicillin failed to kill a small subpopulation of the cells. Despite the discovery many decades ago, the specific mechanisms of Staphylococcus aureus persistence is largely unknown. Recently renewed interest has emerged due to the rise of chronic infections caused by pathogens such as M. tuberculosis, B. burgdorferi, S. aureus, P. aeruginosa, and E. coli. Treatments for chronic infections are lacking and antibiotic resistance is becoming a bigger issue. The goal of our research is to define the mechanisms involved in S. aureus persistence and cell death to improve our knowledge of genes and molecular pathways that can be used as targets for drugs to eradicate chronic infections. -

2016 Medicines in Development for Rare Diseases a LIST of ORPHAN DRUGS in the PIPELINE
2016 Medicines in Development for Rare Diseases A LIST OF ORPHAN DRUGS IN THE PIPELINE Autoimmune Diseases Product Name Sponsor Official FDA Designation* Development Status Actemra® Genentech treatment of systemic sclerosis Phase III tocilizumab South San Francisco, CA www.gene.com Adempas® Bayer HealthCare Pharmaceuticals treatment of systemic sclerosis Phase II riociguat Whippany, NJ www.pharma.bayer.com ARA 290 Araim Pharmaceuticals treatment of neuropathic pain in patients Phase II Tarrytown, NY with sarcoidosis www.ariampharma.com ARG201 arGentis Pharmaceuticals treatment of diffuse systemic sclerosis Phase II (type 1 native bovine skin Collierville, TN www.argentisrx.com collagen) BYM338 Novartis Pharmaceuticals treatment of inclusion body myositis Phase III (bimagrumab) East Hanover, NJ www.novartis.com CCX168 ChemoCentryx treatment of anti-neutrophil cytoplasmic Phase II (5a receptor antagonist) Mountain View, CA auto-antibodies associated vasculitides www.chemocentryx.com (granulomatosis with polyangitis or Wegener's granulomatosis), microscopic polyangitis, and Churg-Strauss syndrome * This designation is issued by the FDA's Office of Orphan Products Development while the drug is still in development. The designation makes the sponsor of the drug eligible for entitlements under the Orphan Drug Act of 1983. The entitlements include seven years of marketing exclusivity following FDA approval of the drug for the designated use. Medicines in Development: Rare Diseases | 2016 1 Autoimmune Diseases Product Name Sponsor Official FDA -
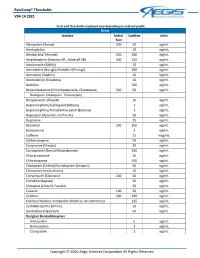
Paincomp® Thresholds V04.14.2021 Copyright © 2020 Aegis Sciences
PainComp® Thresholds V04.14.2021 Tests and thresholds employed vary depending on ordered profile Urine Analyte Initial Confirm Units Test Alprazolam (Xanax) 200 50 ng/mL Amitriptyline 10 ng/mL Amobarbital (Amytal) 200 200 ng/mL Amphetamine (Adzenys ER , Adderall XR) 500 250 ng/mL Aripiprazole (Abilify) 10 ng/mL Armodafinil (Nuvigil)/Modafinil (Provigil) 100 ng/mL Asenapine (Saphris) 10 ng/mL Atomoxetine (Strattera) 10 ng/mL Baclofen 500 ng/mL Benzodiazepines (Chlordiazepoxide, Clorazepate, 200 50 ng/mL Diazepam, Oxazepam, Temazepam) Brexpiprazole (Rexulti) 10 ng/mL Buprenorphine/Sublingual (Belbuca) 1 ng/mL Buprenorphine/Transdermal patch (Butrans) 1 ng/mL Bupropion (Aplenzin, Forfivo XL) 50 ng/mL Buspirone 25 ng/mL Butalbital 200 200 ng/mL Butorphanol 1 ng/mL Caffeine 15 mcg/mL Carbamazepine 50 ng/mL Cariprazine (Vraylar) 20 ng/mL Carisoprodol (Soma)/Meprobamate 200 ng/mL Chlorpromazine 10 ng/mL Chlorzoxazone 500 ng/mL Citalopram (Celexa)/Escitalopram (Lexapro) 50 ng/mL Clomipramine (Anafranil) 10 ng/mL Clonazepam (Klonopin) 200 50 ng/mL Clonidine (Kapvay) 50 ng/mL Clozapine (Clozaril, Fazaclo) 50 ng/mL Cocaine 100 50 ng/mL Codeine 200 100 ng/mL Cotinine/Nicotine metabolite (Habitrol, Nicoderm CQ) 125 ng/mL Cyclobenzaprine (Amrix) 10 ng/mL Dantrolene (Dantrium) 50 ng/mL Designer Benzodiazepines Adinazolam 5 ng/mL Bromazolam 1 ng/mL Clonazolam 1 ng/mL Copyright © 2020 Aegis Sciences Corporation All Rights Reserved PainComp® Thresholds V04.14.2021 Deschloroetizolam 1 ng/mL Diclazepam 1 ng/mL Etizolam 1 ng/mL Flualprazolam 1 ng/mL Flubromazepam -

Bulk Drug Substances Nominated for Use in Compounding Under Section 503B of the Federal Food, Drug, and Cosmetic Act
Updated June 07, 2021 Bulk Drug Substances Nominated for Use in Compounding Under Section 503B of the Federal Food, Drug, and Cosmetic Act Three categories of bulk drug substances: • Category 1: Bulk Drug Substances Under Evaluation • Category 2: Bulk Drug Substances that Raise Significant Safety Risks • Category 3: Bulk Drug Substances Nominated Without Adequate Support Updates to Categories of Substances Nominated for the 503B Bulk Drug Substances List1 • Add the following entry to category 2 due to serious safety concerns of mutagenicity, cytotoxicity, and possible carcinogenicity when quinacrine hydrochloride is used for intrauterine administration for non- surgical female sterilization: 2,3 o Quinacrine Hydrochloride for intrauterine administration • Revision to category 1 for clarity: o Modify the entry for “Quinacrine Hydrochloride” to “Quinacrine Hydrochloride (except for intrauterine administration).” • Revision to category 1 to correct a substance name error: o Correct the error in the substance name “DHEA (dehydroepiandosterone)” to “DHEA (dehydroepiandrosterone).” 1 For the purposes of the substance names in the categories, hydrated forms of the substance are included in the scope of the substance name. 2 Quinacrine HCl was previously reviewed in 2016 as part of FDA’s consideration of this bulk drug substance for inclusion on the 503A Bulks List. As part of this review, the Division of Bone, Reproductive and Urologic Products (DBRUP), now the Division of Urology, Obstetrics and Gynecology (DUOG), evaluated the nomination of quinacrine for intrauterine administration for non-surgical female sterilization and recommended that quinacrine should not be included on the 503A Bulks List for this use. This recommendation was based on the lack of information on efficacy comparable to other available methods of female sterilization and serious safety concerns of mutagenicity, cytotoxicity and possible carcinogenicity in use of quinacrine for this indication and route of administration. -

Eradication of Bacterial Persisters with Antibiotic-Generated Hydroxyl Radicals
Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals Sarah Schmidt Grant a,b,c,1, Benjamin B. Kaufmanna,c,d,1, Nikhilesh S. Chandd,e, Nathan Haseleya,d,f, and Deborah T. Hunga,b,c,d,2 aBroad Institute of MIT and Harvard, Cambridge, MA 02142; bDivision of Pulmonary and Critical Care Medicine, Department of Medicine, Brigham and Women’s Hospital, Boston, MA 02114; cDepartment of Molecular Biology and Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, MA 02114; dDepartment of Microbiology and Immunobiology, Harvard Medical School, Boston, MA 02115; eDepartment of Molecular and Cellular Biology, Harvard University, Cambridge, MA 02138; and fHarvard–MIT Division of Health Sciences and Technology, Cambridge, MA 02139 Edited by* Eric S. Lander, Broad Institute of MIT and Harvard, Cambridge, MA, and approved June 11, 2012 (received for review March 2, 2012) During Mycobacterium tuberculosis infection, a population of bac- terial cell numbers but do not sterilize the mouse (8). A plateau teria likely becomes refractory to antibiotic killing in the absence of is typically reached during which numbers of viable bacteria genotypic resistance, making treatment challenging. We describe stabilize. In addition to the mouse infection model, the inability an in vitro model capable of yielding a phenotypically antibi- to sterilize has been observed in the zebra fish (Mycobacterium otic-tolerant subpopulation of cells, often called persisters, within marinum), guinea pig (M. tuberculosis), and macrophage populations of Mycobacterium smegmatis and M. tuberculosis.We (M. tuberculosis) infection models (9–11). In vitro, the survival find that persisters are distinct from the larger antibiotic-suscepti- of a similar small subpopulation can also be observed when ble population, as a small drop in dissolved oxygen (DO) satura- a culture is exposed to high doses of antibiotics (12, 13).