Influence of Additives on Ethylcellulose Coatings
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CHEMICAL RESISTANCE CHART Chemical Resistance Glove Chart
CHEMICAL RESISTANCE CHART Chemical Resistance Glove Chart P = Poor | F = Fair | G = Good | E = Excellent | X = Not Tested Chemical Latex Nitrile 1,1,1,2-Tetrachloroethane P F P 1,1,1-Trichloroethane P P P 1,1,2-Trichloroethane P P P 1,2,4,5-Tetrachlorobenzene X E P 1,2,4-Trichlorobenzene P F P 1,2-Dichlorobenzene (o-Dichlorobenzene) P P P 1,2-Dichloroethylene P P P 1,2-Propylenoxide P P P 1,3-Dioxane F P P 1,4-Dioxane P P P 1-Nitropropane G P P 2-(Dietylamino)ethanol P E P 2-Chloroethanol X P P 2-Hydroxyethyl acrylate P P P 2-Hydroxyethyl methacrylate (HEMA) X X X 2-Nitropropane P P P 2-Nitrotoluene P P P 3-Bromopropionic acid G F X 4,4-Methylenedianiline (MDA) X X X 5-Fluorouracil X X X Acetaldehyde G P P Acetamide G P P Acetate solvent G F P Acetic acid (glacial acetic acid) 99+ % G F F Acetic acid 30% E G F Acetic acid anhydride G F P Acetone F P P Acetonitrile F F P Acetyl chloride P P P Acetylene G E F Acryl Acid P F P Acrylamide, 30-70% P E F Acrylic Acid G G P Acrylonitrile P P P Adipic acid E E X Allyl alcohol P P P Allylamine P P P Allylchloride (3-Chloroporpene) P P P Aluminium Acetate E G X Aluminium Chloride E E G Aluminium Fluoride G E F Aluminium Hydroxide P E F Aluminium Nitrate E E X Aluminium Potassium Sulfate E E F Aluminium Sulfate E E G Amine G P Ammonia anhdyrous P G G Ammonia gas (cold) E E X Ammonia gas (hot) P P X Ammonia Nitrate X F X 74 Chemical Resistance Glove Chart P = Poor | F = Fair | G = Good | E = Excellent | X = Not Tested Chemical Latex Nitrile Ammonia solution, 30% P E X Ammonium Acetate E E X Ammonium -

Product and Product Group Discharges Subject to Effluent Limitations and Standards for Organic Chemicals, Plastics, and Syntheti
PRODUCT AND PRODUCT GROUP DISCHARGES SUBJECT TO EFFLUENT LIMITATIONS AND STANDARDS for the ORGANIC CHEMICALS, PLASTICS, AND SYNTHETIC FIBERS POINT SOURCE CATEGORY - 40 CFR 414 April 2005 Office of Water U.S. Environmental Protection Agency 1200 Pennsylvania Ave., N.W. Washington, D.C. 20460 Table of Contents Section Page Introduction. 1 1 Summary of the 40 CFR 414 . .2 1.1 Regulated Parameters. 3 1.2 Requirements for Direct and Indirect Dischargers. 3 2. Identifying and Classifying Products Whose Production May be Subject to the OCPSF Regulation. 5 2.1 The SIC Manual and Codes . 5 2.2 Industrial Categories Applicable to Chemicals and Allied Products. .6 2.3 Part 414 Applicability to Production of Chemicals and Chemical Products. .6 2.4 Applicability of Wastewater from On-Site Auxiliary Operations. .8 2.5 General Discussion of OCPSF-Related Products Whose Manufacture May Not Be Regulated by Part 414 . .8 2.5.1 Products Classified and Previously Reported under Specific SIC Codes That Are Not Subject to Part 414 . .8 2.5.2 Products Listed in Part 414 That Are Regulated by Another Industrial Category Are Not Subject to Part 414 in Certain Circumstances . .10 2.5.2.1 Organic Chemicals Regulated by the Iron and Steel Category (40 CFR 420) . 10 2.5.2.2 Organic Chemicals Regulated by the Pesticides Chemicals Category (40 CFR 455). 11 2.5.2.3 Organic Chemicals Regulated by the Pharmaceutical Category. 11 2.5.2.4 Products Regulated by the Plastics Molding & Forming Category (40 CFR 463). 12 2.5.2.5 Organic Chemicals Regulated by the Soap and Detergent Category (40 CFR 417). -
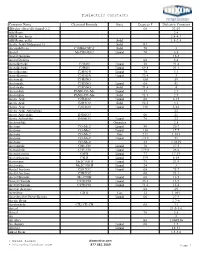
Dielectric Constants
Dielectric Constants Common Name Chemical Formula State Degrees F Dielectric Constant (Ethylenedioxy)diethanol-2,2` 68 23.69 ABS Resin 2.4 ABS Resin, lump 2.4-4.1 ABS Resin, pellet Solid 1.5-2.5 Acatic.Acid (36degrees F) Solid 4.1 Acenaphthene C10H6(CH2)2 Solid 70 3 Acetal MeCH(OEt)2 Liquid 70 3.6 Acetal Bromide 16.5 Acetal Doxime 68 3.4 Acetaldehyde C2H4O Liquid 50 21.8 Acetaldehyde C2H4O Liquid 69.8 21.1 Acetaldoxime C2H5ON Liquid 70 3.4 Acetaldoxime C2H5ON Liquid 73.4 3 Acetamide C3H5NO 180 59 Acetamide C3H5NO Liquid 68 41 Acetamide C3H5NO Solid 71.6 4 Acetanilide PhNH-CO-Me Liquid 71 2.9 Acetanilide PhNH-CO-Me Solid 71.6 2.9 Acetic Acid C2H4O2 Liquid 68 6.15 Acetic Acid C2H4O2 Solid 32.5 4.1 Acetic Acid C2H4O2 Liquid 158 6.62 Acetic Acid, Anhydrous 22 Acetic Anhydride H4H6O3 66 21 Acetic Anhydride H4H6O3 Liquid 70 22 Acetoanilide Granules 2.8 Acetone CO-Me2 Liquid 80 20.7 Acetone CO-Me2 Liquid 130 17.7 Acetone CO-Me2 Gas 212 1.015 Acetone CO-Me2 Liquid -112 31 Acetone CO-Me2 32 1.0159 Acetonitrile CH3-CN Liquid 70 37.5 Acetonitrile CH3-CN Liquid 179.6 26.6 Acetophenone C8H8 Liquid 77 17.39 Acetophenone C8H8 Liquid 394 8.64 Acetoxime Me2C:NOH Liquid 75 23.9 Acetoxime Me2C:NOH Liquid 24 3 Acetyl Acetone C5H2O2 Liquid 68 25.7 Acetyl Acetone C5H2O2 68 23.1 Acetyl Bromide Me-COBr Liquid 68 16.5 Acetyl Chloride C2H3ClO Liquid 35.6 16.9 Acetyl Chloride C2H3ClO Liquid 71.6 15.8 Acetyle Acetone 68 25 Acetylene C2H2 Gas 32 1.001 Acetylmethyl Hexyl Ketone Liquid 66 27.9 Acrylic Resin 2.7-6 Acrylonitrile CH2:CH-CN 68 33 Acteal 21.0-3.6 Acteal -

Film-Forming Systems for Dermal Drug Delivery
pharmaceutics Review Film-Forming Systems for Dermal Drug Delivery Larissa Carine Pünnel and Dominique Jasmin Lunter * Department of Pharmaceutical Technology, Eberhard Karls University, Auf der Morgenstelle 8, 72076 Tübingen, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-7071-297-4558; Fax: +49-7071-295-531 Abstract: Film-forming formulations represent a novel form of sustained release dermatic products. They are applied to the skin as a liquid or semi-solid preparation. By evaporation of the volatile solvent on the skin, the polymer contained in the formulation forms a solid film. Various film-forming formulations were tested for their water and abrasion resistance and compared with conventional semi-solid formulations. Penetration and permeation studies of the formulations indicate a potential utility as transdermal therapeutic systems. They can be used as an alternative to patch systems to administer a variety of drugs in a topical way and may provide sustained release characteristics. Keywords: film-forming system; topical drug delivery; polymeric formulation 1. Introduction 1.1. Skin The skin is the largest organ of the human body [1]. It acts as a protective barrier against external influences such as ultraviolet radiation, chemical and physical insults, attacks by harmful microorganisms, and mechanical irritation. Furthermore, the skin regu- Citation: Pünnel, L.C.; Lunter, D.J. lates physiological parameters of the body by providing a barrier against water evaporation Film-Forming Systems for Dermal and temperature loss [2]. Drug Delivery. Pharmaceutics 2021, 13, Skin diseases such as infections triggered by bacteria or viruses, as well as the immuno- 932. -

Basic Concepts of Cellulose Polymers - a Comprehensive Review
Archives of Pharmacy Practice Review Article ISSN 2045-080X Vol 3 , Issue 3 , 2012 Basic Concepts Of Cellulose Polymers - A Comprehensive Review Harika K, Sunitha K, Pavan Kumar P, Maheshwar K and Madhusudan Rao Y* Department of Pharmaceutics, National facilities in Engineering and Technology with Industrial Collaboration (NAFETIC) centre, University College of Pharmaceutical Sciences, Kakatiya University, Warangal – 506 009, A.P. India. Citation: Harika K, Sunitha K, Pavan Kumar P, Maheshwar K and Madhusudan Rao Y. Basic Concepts Of Cellulose Polymers- A Comprehensive Review. Archives of Pharmacy Practice. 2012; 3(3)pp202-216. Abstract Man’s pursuance for new and improved materials has been should promote innovation. To assist the exploitation expanding with time and it can be said that it is unending. of novel drug delivery systems the need for polymers Though introduced very late in the chain of materials, continues to increase. polymers occupy a major place and pivotal position in our This review serves as a comprehensive source to improve materials map today. Unfolding of the science of polymers and understanding of cellulose derivative polymers and create polymer – based materials had evoked lot of interest and made new avenues in development of a delivery system. In them as a class of materials for their potential use in the field of addition, this review presents in-depth information on pharmaceuticals and industry based products. In recent years, various aspects of polymer chemistry, nomenclature, an awareness and understanding of these polymers has various polymer grades, physical characteristics of increased based upon the following factors. polymers, solubility, and the utility of polymers for As pharmaceutically active ingredients continue to become various drug delivery systems.