Short Or Long Interval Between Priming and Boosting: Does It Impact on the Vaccine Immunogenicity?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Systematic Booster After Rabies Pre-Exposure Prophylaxis to Alleviate Rabies Antibody Monitoring in Individuals at Risk of Occupational Exposure
Article Systematic Booster after Rabies Pre-Exposure Prophylaxis to Alleviate Rabies Antibody Monitoring in Individuals at Risk of Occupational Exposure Perrine Parize 1,*, Jérémie Sommé 2 , Laura Schaeffer 3, Florence Ribadeau-Dumas 1, Sheherazade Benabdelkader 1, Agnès Durand 4, Arnaud Tarantola 1, Johann Cailhol 5, Julia Goesch 5, Lauriane Kergoat 1 , Anne-Sophie Le Guern 6, Marie-Laurence Mousel 2, Laurent Dacheux 1 , Paul-Henri Consigny 5, Arnaud Fontanet 3,7, Beata Francuz 2 and Hervé Bourhy 1 1 Institut Pasteur, Unit Lyssavirus Epidemiology and Neuropathology, National Reference Center for Rabies and WHO Collaborating Centre for Reference and Research on Rabies, 75015 Paris, France; fl[email protected] (F.R.-D.); [email protected] (S.B.); [email protected] (A.T.); [email protected] (L.K.); [email protected] (L.D.); [email protected] (H.B.) 2 Institut Pasteur, Occupational Health Department, 75015 Paris, France; [email protected] (J.S.); [email protected] (M.-L.M.); [email protected] (B.F.) 3 Institut Pasteur, Emerging Diseases Epidemiology Unit, Centre for Global Health Research and Education, 75015 Paris, France; [email protected] (L.S.); [email protected] (A.F.) 4 Laboratoire Cerballiance, 75017 Paris, France; [email protected] 5 Citation: Parize, P.; Sommé, J.; Institut Pasteur, Centre Médical, Centre d’Infectiologie Necker-Pasteur, 75015 Paris, France; Schaeffer, L.; Ribadeau-Dumas, F.; [email protected] (J.C.); [email protected] (J.G.); [email protected] (P.-H.C.) 6 Benabdelkader, S.; Durand, A.; Institut Pasteur, Laboratory of the Medical Center, 75015 Paris, France; [email protected] 7 Conservatoire National des Arts et Métiers, 75003 Paris, France Tarantola, A.; Cailhol, J.; Goesch, J.; * Correspondence: [email protected]; Tel.: +33-140-613-436 Kergoat, L.; et al. -

COVID-19 Vaccination Programme: Information for Healthcare Practitioners
COVID-19 vaccination programme Information for healthcare practitioners Republished 6 August 2021 Version 3.10 1 COVID-19 vaccination programme: Information for healthcare practitioners Document information This document was originally published provisionally, ahead of authorisation of any COVID-19 vaccine in the UK, to provide information to those involved in the COVID-19 national vaccination programme before it began in December 2020. Following authorisation for temporary supply by the UK Department of Health and Social Care and the Medicines and Healthcare products Regulatory Agency being given to the COVID-19 Vaccine Pfizer BioNTech on 2 December 2020, the COVID-19 Vaccine AstraZeneca on 30 December 2020 and the COVID-19 Vaccine Moderna on 8 January 2021, this document has been updated to provide specific information about the storage and preparation of these vaccines. Information about any other COVID-19 vaccines which are given regulatory approval will be added when this occurs. The information in this document was correct at time of publication. As COVID-19 is an evolving disease, much is still being learned about both the disease and the vaccines which have been developed to prevent it. For this reason, some information may change. Updates will be made to this document as new information becomes available. Please use the online version to ensure you are accessing the latest version. 2 COVID-19 vaccination programme: Information for healthcare practitioners Document revision information Version Details Date number 1.0 Document created 27 November 2020 2.0 Vaccine specific information about the COVID-19 mRNA 4 Vaccine BNT162b2 (Pfizer BioNTech) added December 2020 2.1 1. -

Considerations for Causality Assessment of Neurological And
Occasional essay J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp-2021-326924 on 6 August 2021. Downloaded from Considerations for causality assessment of neurological and neuropsychiatric complications of SARS- CoV-2 vaccines: from cerebral venous sinus thrombosis to functional neurological disorder Matt Butler ,1 Arina Tamborska,2,3 Greta K Wood,2,3 Mark Ellul,4 Rhys H Thomas,5,6 Ian Galea ,7 Sarah Pett,8 Bhagteshwar Singh,3 Tom Solomon,4 Thomas Arthur Pollak,9 Benedict D Michael,2,3 Timothy R Nicholson10 For numbered affiliations see INTRODUCTION More severe potential adverse effects in the open- end of article. The scientific community rapidly responded to label phase of vaccine roll- outs are being collected the COVID-19 pandemic by developing novel through national surveillance systems. In the USA, Correspondence to SARS- CoV-2 vaccines (table 1). As of early June Dr Timothy R Nicholson, King’s roughly 372 adverse events have been reported per College London, London WC2R 2021, an estimated 2 billion doses have been million doses, which is a lower rate than expected 1 2LS, UK; timothy. nicholson@ administered worldwide. Neurological adverse based on the clinical trials.6 kcl. ac. uk events following immunisation (AEFI), such as In the UK, adverse events are reported via the cerebral venous sinus thrombosis and demyelin- MB and AT are joint first Coronavirus Yellow Card reporting website. As of ating episodes, have been reported. In some coun- authors. early June 2021, approximately 250 000 Yellow tries, these have led to the temporary halting of BDM and TRN are joint senior Cards have been submitted, equating to around authors. -

Priming of Virus-Immune Memory T Cells in Newborn Mice DAVID H
INFECTION AND IMMUNITY, Jan. 1984, p. 202-205 Vol. 43, No. 1 0019-9567/84/010202-04$02.00/0 Copyright (© 1984, American Society for Microbiology Priming of Virus-Immune Memory T Cells in Newborn Mice DAVID H. SCHWARTZ, JULIA L. HURWITZ,* NEIL S. GREENSPAN,t AND PETER C. DOHERTYt The Wistar Institute ofAnatomy and Biology, Philadelphia, Pennsylvania 19104 Received 6 May 1983/Accepted 27 September 1983 Neonatal BALB/c mice can be primed at birth by intravenous inoculation of a small dose of A/Puerto Rico/8/34 (H1N1) (PR8) influenza virus, UV-inactivated PR8 virus, or PR8 virus complexed with monoclonal antibody to give a secondary cytotoxic T lymphocyte response when restimulated in vitro as adults. The frequency of responding T cells after secondary stimulation in vitro is approximately 40% of that found for adult mice primed intraperitoneally with a large dose of PR8 virus. The majority of the T cells generated from mice primed at birth or as adults are cross-reactive for H-2-compatible targets infected with the PR8 (H1N1) or A/Hong Kong/X31 (H3N2) viruses. Splenocytes from neonates receiving UV-inactivated vaccinia virus at birth give an augmented secondary cytotoxic T lymphocyte response when restimulated 8 days later in adoptive irradiated adult hosts. We found no indications of specific immunological unrespon- siveness in mice exposed to either virus. We have analyzed previously the development of virus- cells per ml for 1 h at 37°C. Graded numbers of effector specific T cell competence in neonatal mice and have spleen cells and 2 x 105 syngeneic virus-infected, irradiated reported the presence of vaccinia virus-specific cytotoxic T stimulator cells were distributed in 100-.pl portions to the 60 lymphocyte (CTL) precursors in the thymus of 0- to 3-day- inside wells of 96-well, V-bottom plates. -

Priming with Inflammatory Cytokines Is Not a Prerequisite to Increase
Lange-Consiglio et al. Stem Cell Research & Therapy (2020) 11:99 https://doi.org/10.1186/s13287-020-01611-z RESEARCH Open Access Priming with inflammatory cytokines is not a prerequisite to increase immune- suppressive effects and responsiveness of equine amniotic mesenchymal stromal cells Anna Lange-Consiglio1* , Pietro Romele2, Marta Magatti2, Antonietta Silini2, Antonella Idda1, Nicola Antonio Martino3, Fausto Cremonesi1 and Ornella Parolini2,4 Abstract Background: Equine amniotic mesenchymal stromal cells (AMSCs) and their conditioned medium (CM) were evaluated for their ability to inhibit in vitro proliferation of peripheral blood mononuclear cells (PBMCs) with and without priming. Additionally, AMSC immunogenicity was assessed by expression of MHCI and MHCII and their ability to counteract the in vitro inflammatory process. Methods: Horse PBMC proliferation was induced with phytohemagglutinin. AMSC priming was performed with 10 ng/ml of TNF-α, 100 ng/ml of IFN-γ, and a combination of 5 ng/ml of TNF-α and 50 ng/ml of IFN-γ. The CM generated from naïve unprimed and primed AMSCs was also tested to evaluate its effects on equine endometrial cells in an in vitro inflammatory model induced by LPS. Immunogenicity marker expression (MHCI and II) was evaluated by qRT-PCR and by flow cytometry. Results: Priming does not increase MHCI and II expression. Furthermore, the inhibition of PBMC proliferation was comparable between naïve and conditioned cells, with the exception of AMSCs primed with both TNF-α and IFN-γ that had a reduced capacity to inhibit T cell proliferation. However, AMSC viability was lower after priming than under other experimental conditions. -

Rapid Induction of Antigen-Specific CD4+ T Cells Guides Coordinated Humoral and Cellular Immune Responses to SARS-Cov-2 Mrna Vaccination
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.21.440862; this version posted April 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Rapid induction of antigen-specific CD4+ T cells guides coordinated humoral and cellular immune responses to SARS-CoV-2 mRNA vaccination Authors: Mark M. Painter1,2, †, Divij Mathew1,2, †, Rishi R. Goel1,2, †, Sokratis A. Apostolidis1,2,3, †, Ajinkya Pattekar2, Oliva Kuthuru1, Amy E. Baxter1, Ramin S. Herati4, Derek A. Oldridge1,5, Sigrid Gouma6, Philip Hicks6, Sarah Dysinger6, Kendall A. Lundgreen6, Leticia Kuri-Cervantes1,6, Sharon Adamski2, Amanda Hicks2, Scott Korte2, Josephine R. Giles1,7,8, Madison E. Weirick6, Christopher M. McAllister6, Jeanette Dougherty1, Sherea Long1, Kurt D’Andrea1, Jacob T. Hamilton2,6, Michael R. Betts1,6, Paul Bates6, Scott E. Hensley6, Alba Grifoni9, Daniela Weiskopf9, Alessandro Sette9, Allison R. Greenplate1,2, E. John Wherry1,2,7,8,* Affiliations 1 Institute for Immunology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA 2 Immune Health™, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA 3 Division of Rheumatology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA 4 NYU Langone Vaccine Center, Department of Medicine, New York University School of Medicine, New York, NY 5 Department -

Statements Contained in This Release As the Result of New Information Or Future Events Or Developments
Pfizer and BioNTech Provide Update on Booster Program in Light of the Delta-Variant NEW YORK and MAINZ, GERMANY, July 8, 2021 — As part of Pfizer’s and BioNTech’s continued efforts to stay ahead of the virus causing COVID-19 and circulating mutations, the companies are providing an update on their comprehensive booster strategy. Pfizer and BioNTech have seen encouraging data in the ongoing booster trial of a third dose of the current BNT162b2 vaccine. Initial data from the study demonstrate that a booster dose given 6 months after the second dose has a consistent tolerability profile while eliciting high neutralization titers against the wild type and the Beta variant, which are 5 to 10 times higher than after two primary doses. The companies expect to publish more definitive data soon as well as in a peer-reviewed journal and plan to submit the data to the FDA, EMA and other regulatory authorities in the coming weeks. In addition, data from a recent Nature paper demonstrate that immune sera obtained shortly after dose 2 of the primary two dose series of BNT162b2 have strong neutralization titers against the Delta variant (B.1.617.2 lineage) in laboratory tests. The companies anticipate that a third dose will boost those antibody titers even higher, similar to how the third dose performs for the Beta variant (B.1.351). Pfizer and BioNTech are conducting preclinical and clinical tests to confirm this hypothesis. While Pfizer and BioNTech believe a third dose of BNT162b2 has the potential to preserve the highest levels of protective efficacy against all currently known variants including Delta, the companies are remaining vigilant and are developing an updated version of the Pfizer-BioNTech COVID-19 vaccine that targets the full spike protein of the Delta variant. -

COVID-19 Vaccines Frequently Asked Questions
Page 1 of 12 COVID-19 Vaccines 2020a Frequently Asked Questions Michigan.gov/Coronavirus The information in this document will change frequently as we learn more about COVID-19 vaccines. There is a lot we are learning as the pandemic and COVID-19 vaccines evolve. The approach in Michigan will adapt as we learn more. September 29, 2021. Quick Links What’s new | Why COVID-19 vaccination is important | Booster and additional doses | What to expect when you get vaccinated | Safety of the vaccine | Vaccine distribution/prioritization | Additional vaccine information | Protecting your privacy | Where can I get more information? What’s new − Pfizer booster doses recommended for some people to boost waning immunity six months after completing the Pfizer vaccine. Why COVID-19 vaccination is important − If you are fully vaccinated, you don’t have to quarantine after being exposed to COVID-19, as long as you don’t have symptoms. This means missing less work, school, sports and other activities. − COVID-19 vaccination is the safest way to build protection. COVID-19 is still a threat, especially to people who are unvaccinated. Some people who get COVID-19 can become severely ill, which could result in hospitalization, and some people have ongoing health problems several weeks or even longer after getting infected. Even people who did not have symptoms when they were infected can have these ongoing health problems. − After you are fully vaccinated for COVID-19, you can resume many activities that you did before the pandemic. CDC recommends that fully vaccinated people wear a mask in public indoor settings if they are in an area of substantial or high transmission. -

An Overview of COVID Vaccine Clinical Trial Results & Some Challenges
An overview of COVID vaccine clinical trial results & some challenges DCVMN Webinar December 8th, 2020 Access to COVID-19 tools ACCESSACCESS TO TOCOVID-19 COVID-19 TOOLS TOOLS (ACT) (ACT) ACCELERATOR ACCELERATOR (ACT) accelerator A GlobalA GlobalCollaboration Collaboration to Accelerate tothe AccelerateDevelopment, the Production Development, and Equitable Production Access to New and Equitable AccessCOVID-19 to New diagnostics, COVID-19 therapeutics diagnostics, and vaccines therapeutics and vaccines VACCINES DIAGNOSTICS THERAPEUTICS (COVAX) Development & Manufacturing Led by CEPI, with industry Procurement and delivery at scale Led by Gavi Policy and allocation Led by WHO Key players SOURCE: (ACT) ACCELERATOR Commitment and Call to Action 24th April 2020 ACT-A / COVAX governance COVAX COORDINATION MEETING CEPI Board Co-Chair: Jane Halton Co-Chair: Dr. Ngozi Gavi Board Workstream leads + DCVMN and IFPMA-selected Reps As needed – R&D&M Chair; COVAX IPG Chair Development & Manufacturing Procurement and delivery Policy and allocation (COVAX) at scale Led by (with industry) Led by Led by R&D&M Investment Committee COVAX Independent Product Group Technical Review Group Portfolio Group Vaccine Teams SWAT teams RAG 3 COVAX SWAT teams are being set up as a joint platform to accelerate COVID- 19 Vaccine development and manufacturing by addressing common challenges together Timely and targeted Multilateral Knowledge-based Resource-efficient Addresses specific cross- Establishes a dialogue Identifies and collates Coordinates between developer technical and global joint effort most relevant materials different organizations/ challenges as they are across different COVID-19 and insights across the initiatives to limit raised and/or identified vaccines organizations broader COVID-19 duplications and ensure on an ongoing basis (incl. -
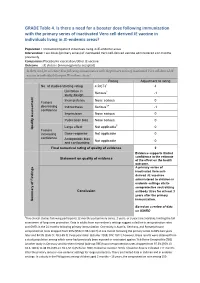
GRADE Table 4. Is There a Need for a Booster Dose Following Immunization Wi
GRADE Table 4. Is there a need for a booster dose following immunization with the primary series of inactivated Vero cell-derived JE vaccine in individuals living in JE-endemic areas? Population : Immunocompetent individuals living in JE-endemic areas Intervention: Two doses (primary series) of inactivated Vero cell-derived vaccine administered ≥12 months previously Comparison: Placebo/no vaccination/other JE vaccine Outcome : JE disease (immunogenicity accepted) Is there need for a booster dose following immunization with the primary series of inactivated Vero cell-derived JE vaccine in individuals living in JE-endemic areas? Rating Adjustment to rating No. of studies/starting rating 4 RCTs1 4 Limitation in 2 Serious -1 study design Inconsistency None serious 0 Factors decreasing Indirectness Serious3,4 -1 confidence Imprecision None serious 0 Publication bias None serious 0 Large effect Not applicable5 0 Quality Assessment Quality Factors increasing Dose-response Not applicable 0 confidence Antagonistic bias Not applicable 0 and confounding Final numerical rating of quality of evidence 2 Evidence supports limited confidence in the estimate Statement on quality of evidence of the effect on the health outcome. A primary series of inactivated Vero cell- derived JE vaccines administered to children in endemic settings elicits seroprotective neutralizing Conclusion antibody titres for at least 3 years after the primary immunization. Summary of Findings of Summary Based on a review of data on IXIARO 1Five clinical studies following participants 12 months post-primary series, 2 years, or 3 years are available, limiting the full assessment of long-term protection. Data in adults from non-endemic settings suggest a decline in seroprotection rates and GMTs in the 24 months following primary immunization. -

Gene-Based Vaccines (GBV)
Advanced Drug Delivery Reviews 170 (2021) 113–141 Contents lists available at ScienceDirect Advanced Drug Delivery Reviews journal homepage: www.elsevier.com/locate/addr Advances in gene-based vaccine platforms to address the COVID-19 pandemic Deborah Pushparajah a,1, Salma Jimenez a,c,1,ShirleyWonga,HibahAlattasa, Nafiseh Nafissi b, Roderick A. Slavcev a,b,c,⁎ a School of Pharmacy, University of Waterloo, 10A Victoria St S, Kitchener N2G 1C5, Canada b Mediphage Bioceuticals, 661 University Avenue, Suite 1300, Toronto, ON, M5G 0B7, Canada c Theraphage, 151 Charles St W Suite # 199, Kitchener, ON, N2G 1H6, Canada article info abstract Article history: The novel betacoronavirus, SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), has spread across Received 1 October 2020 the globe at an unprecedented rate since its first emergence in Wuhan City, China in December 2019. Scientific Received in revised form 23 December 2020 communities around the world have been rigorously working to develop a potent vaccine to combat COVID-19 Accepted 1 January 2021 (coronavirus disease 2019), employing conventional and novel vaccine strategies. Gene-based vaccine platforms Available online 7 January 2021 based on viral vectors, DNA, and RNA, have shown promising results encompassing both humoral and cell-mediated immune responses in previous studies, supporting their implementation for COVID-19 vaccine de- Keywords: Coronavirus velopment. In fact, the U.S. Food and Drug Administration (FDA) recently authorized the emergency use of two COVID-19 RNA-based COVID-19 vaccines. We review current gene-based vaccine candidates proceeding through clinical SARS-CoV-2 trials, including their antigenic targets, delivery vehicles, and route of administration. -

A Media Handbook for HIV Vaccine Trials for Africa Acknowledgements
A Media Handbook for HIV Vaccine Trials for Africa Acknowledgements The Media Handbook for HIV Vaccine Trials for Africa was written by Yinka Adeyemi with guidance and direction from Bunmi Makinwa of the department of Policy, Strategy and Research, Dr Jose Esparza, Dr Saladin Osmanov, Claire Pattou, and Coumba Touré of the World Health Organization (WHO)/Joint United Nations Programme on HIV/AIDS (UNAIDS), HIV Vaccine Initiative. We would like to acknowledge the following individuals for their valuable comments and contributions to this handbook: Dr Alashle Abimiku, Dr Omu Anzala, Dr Carlos Arnaldo, Dr Courtney Batholomew, Janet Frohlich, Dr D. A. Gangakhedar, Dr Rodney Hoff. Patrick Jabani, Bachi Karkaria, Dr Tom LaSalvia, Dr Chewe Luo, Nebat Mbewe, Dr Rosemary Musonda, Binod Mahanty, Dr Roy Mugerwa, Ronaldo Mussauer de Lima, Omololu Falobi, Otula Owuor, Kirk Pereira, Dr John Rwomushana, Mario Scheffer, Jaya Shreedhar, Judith Soal, Dr Prasert Thongcharoen, Kathy Ann Waterman and Victor Zonana. The section on Communication and vaccine trials in Thailand (Appendix 1) is based on a UNAIDS report by Nusara Thaitawat, while that on Communication issues in vaccine trials in Uganda (Appendix 2) is based on a UNAIDS report by Ann Fieldler. The section on Communication and preparations for HIV vaccine trials in Kenya (Appendix 3) is by Otula Owuor. A number of fictitious people and organizations are used for illustrative purposes within the text. Any reference to actual persons or organizations is purely coincidental. UNAIDS/01.05E (English original, February 2001) ISBN 92-9173-021-1 © Joint United Nations Programme on HIV/AIDS The designations employed and the presentation of the (UNAIDS) 2001.