Adaptive Evolution and Demographic History of Norway Spruce
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Department of Planning and Zoning
Department of Planning and Zoning Subject: Howard County Landscape Manual Updates: Recommended Street Tree List (Appendix B) and Recommended Plant List (Appendix C) - Effective July 1, 2010 To: DLD Review Staff Homebuilders Committee From: Kent Sheubrooks, Acting Chief Division of Land Development Date: July 1, 2010 Purpose: The purpose of this policy memorandum is to update the Recommended Plant Lists presently contained in the Landscape Manual. The plant lists were created for the first edition of the Manual in 1993 before information was available about invasive qualities of certain recommended plants contained in those lists (Norway Maple, Bradford Pear, etc.). Additionally, diseases and pests have made some other plants undesirable (Ash, Austrian Pine, etc.). The Howard County General Plan 2000 and subsequent environmental and community planning publications such as the Route 1 and Route 40 Manuals and the Green Neighborhood Design Guidelines have promoted the desirability of using native plants in landscape plantings. Therefore, this policy seeks to update the Recommended Plant Lists by identifying invasive plant species and disease or pest ridden plants for their removal and prohibition from further planting in Howard County and to add other available native plants which have desirable characteristics for street tree or general landscape use for inclusion on the Recommended Plant Lists. Please note that a comprehensive review of the street tree and landscape tree lists were conducted for the purpose of this update, however, only -
Estimating Numbers of Embryonic Lethals in Conifers
Heredity 69(1992)308—314 Received 26 November 1991 OThe Genetical Society of Great Britain Estimating numbers of embryonic lethals in conifers OUTI SAVOLAINEN, KATRI KARKKAINEN & HELMI KUITTINEN* Department of Genetics, University of Oulu, Oulu, Fin/and and *Department of Genetics, University of Helsinki, He/sink,, Fin/and Conifershave recessive lethal genes that eliminate most selfed embryos during seed development. It has been estimated that Scots pine has, on average, nine recessive lethals which act during seed development. Such high numbers are not consistent with the level of outcrossing, about 0.9—0.95, which has been observed in natural populations. Correcting for environmental mortality or using partial selfings provides significantly lower estimates of lethals. A similar discordance with numbers of lethals and observed outcrossing rates is true for other species. Keywords:embryoniclethals, inbreeding depression, outcrossing, Pinus sylvestris, Picea omorika. Introduction Reproduction system of conifers Conifershave no self-incompatibility mechanisms but Theproportion of self-pollination in conifers is early-acting inbreeding depression eliminates selfed variable. Sarvas (1962) suggested an average of 26 per embryos before seed maturation (Sarvas, 1962; cent for Pinus sylvestris, while Koski (1970) estimated Hagman & Mikkola, 1963). A genetic model for this values of self-fertilization around 10 per cent. The inbreeding depression has been developed by Koski genera Pinus and Picea have polyzygotic poly- (1971) and Bramlett & Popham (1971). Koski (1971, embryony, i.e. the ovules contain several archegonia. In 1973) has estimated that Pinus sylvestris and Picea Pinus sylvestris, the most common number of arche- abies have on average nine and 10 recessive lethals, gonia is two but it can range from one to five. -
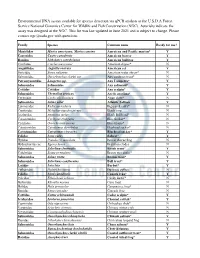
Edna Assay Development
Environmental DNA assays available for species detection via qPCR analysis at the U.S.D.A Forest Service National Genomics Center for Wildlife and Fish Conservation (NGC). Asterisks indicate the assay was designed at the NGC. This list was last updated in June 2021 and is subject to change. Please contact [email protected] with questions. Family Species Common name Ready for use? Mustelidae Martes americana, Martes caurina American and Pacific marten* Y Castoridae Castor canadensis American beaver Y Ranidae Lithobates catesbeianus American bullfrog Y Cinclidae Cinclus mexicanus American dipper* N Anguillidae Anguilla rostrata American eel Y Soricidae Sorex palustris American water shrew* N Salmonidae Oncorhynchus clarkii ssp Any cutthroat trout* N Petromyzontidae Lampetra spp. Any Lampetra* Y Salmonidae Salmonidae Any salmonid* Y Cottidae Cottidae Any sculpin* Y Salmonidae Thymallus arcticus Arctic grayling* Y Cyrenidae Corbicula fluminea Asian clam* N Salmonidae Salmo salar Atlantic Salmon Y Lymnaeidae Radix auricularia Big-eared radix* N Cyprinidae Mylopharyngodon piceus Black carp N Ictaluridae Ameiurus melas Black Bullhead* N Catostomidae Cycleptus elongatus Blue Sucker* N Cichlidae Oreochromis aureus Blue tilapia* N Catostomidae Catostomus discobolus Bluehead sucker* N Catostomidae Catostomus virescens Bluehead sucker* Y Felidae Lynx rufus Bobcat* Y Hylidae Pseudocris maculata Boreal chorus frog N Hydrocharitaceae Egeria densa Brazilian elodea N Salmonidae Salvelinus fontinalis Brook trout* Y Colubridae Boiga irregularis Brown tree snake* -

EASTERN SPRUCE an American Wood
pÍ4 .4 . 1j%4 Mk ,m q ' ' 5 -w +*' 1iiL' r4i41. a')' q I Figure 1.-Range of white spruce (Picea glauca). 506612 2 EASTERN SPRUCE an American wood M.D. Ostrander' DISTRIBUTION monly found in cold, poorly-drained bogs throughout its range, and under such conditions forms dense slow- (Moench) Three spruces-white (Picea glauca growing pure stands. Through most of its range black Voss) , black (Picea , mariana (Mill.) B.S.P.) and red spruce occurs at elevations between 500 and 2,500 Picea rubens collectively known east- ( Sarg.)-are as feet, although substantial stands occur at lower eleva- em spruce. The natural range of white spruce ex- tions in eastern Canada. In the Canadian Rockies and tends from Newfoundland and Labrador west across Alaska it grows up the slopes to the limits of tree Canada to northwestern Alaska and southward growth. It usually is found in pure stands and in mix- through northern New England, New York, Mich- tures with other lowland softwoods, aspen, and birch. igan, Wisconsin, and Minnesota (fig. In western i ) . The natural range of red spruce extends from the mountains in Canada it is found east of coastal Nova Scotia and New Brunswick in Canada, southward British Columbia. Outlying stands also occur in the through New England and eastern New York. It con- Black Hills of South Dakota and in Montana and tinues southward at high elevations in the Appala. Wyoming. White spruce grows under a variety of chian Mountains to western North Carolina and east- climatic conditions, ranging from the wet insular cli- em Tennessee (fig. -

Growth Pattern of Picea Rubens Prior to Canopy Recruitment
pr7 7 AL Plant Ecology 140: 245-253, 1999. 245 rim © 1999 Kluwer Academic Publishers. Printed in the Netherlands. Growth pattern of Picea rubens prior to canopy recruitment Xinyuan Wu l , J. Frank McCormick2 Richard T. Busing3 1 Department of Rangeland Ecology and Management Texas AM University, College Station, TX 77843, USA; 2Department of Ecology and Evolutionary Biology, The University of Tennessee Knoxville, TN 37996, USA; 3Forestry Sciences Laboratory 3200 SW Jefferson Way Corvallis, OR 97331, USA Received 22 July 1997; accepted in revised form 6 October 1998 Key words: Canopy disturbance regime, Great Smoky Mountains, Radial growth, Red spruce, Suppression and release Abstract A majority (72%) of Picea rubens Sarg. (red spruce) trees in an old-growth spruce-fir forest in the Great Smoky Mountains underwent episodes of radial growth suppression and release before they reached the forest canopy. Prior to canopy recruitment, trees experienced an average of 1.43 and a maximum of 7 suppression periods with an average ring width of 0.257 mm. Duration of suppression periods ranged from 4 to 79 years with an average of 19.05 years, which was significantly shorter than the average duration of release periods (29.00 years). Mean ring width in a suppression period was negatively correlated with duration of the suppression period. The opposite was true for release periods. The severity of suppression had no significant effect on mean ring width in subsequent release periods. Greater suppression was observed in the recent growth pattern of current non-canopy trees than in the historical growth pattern reconstructed from current canopy trees. -

Identification of Conifer Trees in Iowa This Publication Is Designed to Help Identify the Most Common Trees Found in Iowa
Identification of Conifer Trees in Iowa This publication is designed to help identify the most common trees found in Iowa. It is based on vegetative characteristics including leaves, fruit, and bark. It is neither complete nor without possible oversights. Separate species are grouped by similar characteristics, mainly based on type and arrangement of leaves. These groups are; awl- or scale- like needles; single needles, flattened with rounded tips; single needles, square in cross section, with pointed tips; and needles in bundles or fasticles of two or more. Remember, vegetative character- istics are quite variable; use more than one specimen for comparison. Awl- or scale-like needles Juniperus Virginiana Eastern Red Cedar Leaves are dark green; leaves are both awl- and scale-like; cone is dark blue and berry-like. Thuja occidentalis Northern White Cedar Leaves are flattened and only of the scale type; cones have 4-6 scales; foliage is light green. Juniperus communis Common Juniper Leaves are awl shaped; cone is dark blue and berry-like. Pm-1383 | May 1996 Single needles, flattened with rounded tips Pseudotsuga menziesii Douglas Fir Needles occur on raised pegs; 3/4-11/4 inches in length; cones have 3-pointed bracts between the cone scales. Abies balsamea Abies concolor Balsam Fir White (Concolor) Fir Needles are blunt and notched at Needles are somewhat pointed, the tip; 3/4-11/2 inches in length. curved towards the branch top and 11/2-3 inches in length; silver green in color. Single needles, Picea abies Norway Spruce square in cross Needles are 1/2-1 inch long; section, with needles are dark green; foliage appears to droop or weep; cone pointed tips is 4-7 inches long. -

Picea Rubens Sarg. Family: Pinaceae Red Spruce
Picea rubens Sarg. Family: Pinaceae Red Spruce The genus Picea is composed of about 30 species native to North America [12] and Eurasia [20]. The word picea comes from the ancient Latin name (pix, picis = pitch) of a pitchy pine, probably Scotch pine (Pinus sylvestris L.). The word rubens means reddish, referring to the reddish brown cones. Other Common Names: Abetina rossa, Adirondack spruce, black spruce, blue spruce, Canadese rode spar, Canadian red spruce, Canadian spruce, double spruce, eastern spruce, epicea rouge du Canada, he balsam, he- balsam, Hudson-fichte, kanadensisk rod-gran, North American red spruce, picea roja de Canada, picea rossa del Canada, red spruce, rot-fichte, sapinette rouge du Canada, spruce pine, spruces d'america, West Virginia spruce, yellow spruce. Distribution: Red spruce is native to Cape Breton Islands, Nova Scotia and New Brunswick, west to Maine, southern Quebec and southeastern Ontario and south to central New York, northeastern Pennsylvania, northern New Jersey and Massachusetts. It also grows in the Appalachian Mountains of extreme western Maryland, eastern West Virginia, northern and western Virginia, western North Carolina and eastern Tennessee. The Tree: Red spruce can reach heights of 110 feet, with diameters of 4.5 feet. At the northern limit of its range, red spruce reaches heights of only 80 feet and diameters of 2 feet. General Wood Characteristics: The wood dries easily and is stable after drying, is moderately light in weight and easily worked, has moderate shrinkage, and is moderately strong, stiff, tough, and hard. It is not very resistant to bending or end-wise compression. -

The Anatomy of Spruce Needles '
THE ANATOMY OF SPRUCE NEEDLES ' By HERBERT F. MARCO 2 Junior forester. Northeastern Forest Experiment Station,^ Forest Service, United States Department of Agriculture INTRODUCTION In 1865 Thomas (16) * made a comparative study of the anatomy of conifer leaves and fomid that the structural variations exhibited by the different species warranted taxonomic considération. Since that time leaf anatomy has become a fertile and interesting field of research. Nearly all genera of gymnosperms have received some attention, and the literature on this subject has become voluminous. A detailed review of the literature will not be attempted in this paper, since com- prehensive reviews have already been published by Fulling (6) and Lacassagne (11). FuUing's paper contains in addition an extensive bibliography on conifer leaf anatomy. Most workers in tMs field of research have confined their efforts to the study of the cross sections of needles. This is partly because longitudinal sections are difficult to obtain and partly because they present but little structural variation of value for identification. The workers who have studied both longitudinal and cross sections have restricted their descrii)tions of longitudinal sections either to specific tissues or to a few species of a large number of genera, and the descrip- tions, although comprehensive, leave much to be desired from the standpoint of detailed information and illustration. Domer (S) was perhaps the first to use sketches to augment keys to and descriptions of the native firs and spruces. His diagrammatic sketches portray the shape of the needles in cross section and the position of the resin canals. Durrell (4) went a step further and illustrated his notes on the North American conifers by camera-lucida drawing^ depicting the orientation and arrangement of the various needle tissues in cross section. -

Measuring Restoration Success CASRI Partnership VIRTUAL Conference 2020
Measuring Restoration Success CASRI Partnership VIRTUAL Conference 2020 November 4 – 5, 2020 CASRI's 2020 "Measuring Restoration Success" conference will focus on quantitative and qualitative measures of success for restoration of the red spruce-northern hardwood ecosystem in Central Appalachia. Decades of boots-on-the-ground restoration actions and associated research and monitoring has resulted in quantifiable success stories and lessons learned, all of which provide important knowledge to inform future actions and approaches. This conference gathers managers, practitioners, scientists, and leaders in the field to discuss the latest research findings, problem-solve common management challenges, and network to advance new and emerging partnerships. Strong partnerships will enable the network to continue to advance landscape resilience and connectivity of red spruce forests across the region. SCHEDULE Wednesday November 4, 2020 --- ZOOM link Day 1 Time Topic Speaker 9:00 – 9:10 Welcome and housekeeping Katy Barlow 9:10 – 9:50 Keynote Presentation: Are We Hitting the Mark Jamie Schuler for Red Spruce Restoration? Session 1: History of CASRI 10:00 – 10:15 The Blister Swamp Conservation and Alton Byers Restoration Project Twenty Years Later 10:15 – 10:30 Red spruce in West Virginia: Early Reports James Rentch from A. D. Hopkins and the W.Va. Agricultural Experiment Station 10:30 – 10:45 Session 1 Discussion Panel Session 2: Soils and Vegetation 11:00 – 11:10 Informed Red Spruce Restoration and James Leonard Conservation Planning using Soil -
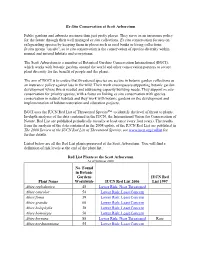
IUCN Red List of Threatened Species™ to Identify the Level of Threat to Plants
Ex-Situ Conservation at Scott Arboretum Public gardens and arboreta are more than just pretty places. They serve as an insurance policy for the future through their well managed ex situ collections. Ex situ conservation focuses on safeguarding species by keeping them in places such as seed banks or living collections. In situ means "on site", so in situ conservation is the conservation of species diversity within normal and natural habitats and ecosystems. The Scott Arboretum is a member of Botanical Gardens Conservation International (BGCI), which works with botanic gardens around the world and other conservation partners to secure plant diversity for the benefit of people and the planet. The aim of BGCI is to ensure that threatened species are secure in botanic garden collections as an insurance policy against loss in the wild. Their work encompasses supporting botanic garden development where this is needed and addressing capacity building needs. They support ex situ conservation for priority species, with a focus on linking ex situ conservation with species conservation in natural habitats and they work with botanic gardens on the development and implementation of habitat restoration and education projects. BGCI uses the IUCN Red List of Threatened Species™ to identify the level of threat to plants. In-depth analyses of the data contained in the IUCN, the International Union for Conservation of Nature, Red List are published periodically (usually at least once every four years). The results from the analysis of the data contained in the 2008 update of the IUCN Red List are published in The 2008 Review of the IUCN Red List of Threatened Species; see www.iucn.org/redlist for further details. -

Apparent Hybridisation of Firecrest and Goldcrest F
Apparent hybridisation of Firecrest and Goldcrest F. K. Cobb From 20th to 29th June 1974, a male Firecrest Regulus ignicapillus was seen regularly, singing strongly but evidently without a mate, in a wood in east Suffolk. The area had not been visited for some time before 20th June, so that it is not known how long he had been present. The wood covers some 10 ha and is mainly deciduous, com prised of oaks Quercus robur, sycamores Acer pseudoplatanus, and silver birches Betula pendula; there is also, however, a scatter of European larches Larix decidua and Scots pines Pinus sylvestris, with an occasional Norway spruce Picea abies. Apart from the silver birches, most are mature trees. The Firecrest sang usually from any one of about a dozen Scots pines scattered over half to three-quarters of a hectare. There was also a single Norway spruce some 18-20 metres high in this area, which was sometimes used as a song post, but the bird showed no preference for it over the Scots pines. He fed mainly in the surround ing deciduous trees, but was never heard to sing from them. The possibility of an incubating female was considered, but, as the male showed no preference for any particular tree, this was thought unlikely. Then, on 30th June, G. J. Jobson saw the Firecrest with another Regulus in the Norway spruce and, later that day, D. J. Pearson and J. G. Rolfe watched this second bird carrying a feather in the same tree. No one obtained good views of it, but, not unnaturally, all assumed that it was a female Firecrest. -

Germination and Early Survival of Picea Rubens Sargent in Experimental Laboratory and Field Plantings
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 3-1965 Germination and Early Survival of Picea rubens Sargent in Experimental Laboratory and Field Plantings Nancy C. Wilson University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Science and Mathematics Education Commons Recommended Citation Wilson, Nancy C., "Germination and Early Survival of Picea rubens Sargent in Experimental Laboratory and Field Plantings. " Master's Thesis, University of Tennessee, 1965. https://trace.tennessee.edu/utk_gradthes/3256 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Nancy C. Wilson entitled "Germination and Early Survival of Picea rubens Sargent in Experimental Laboratory and Field Plantings." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Education. W. W. Wyatt, Major Professor We have read this thesis and recommend its acceptance: H. R. DeSelem, Fred Norris Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) March 1, 1965 To the Graduate Council: I am submitting herewith a thesis written by Nancy c.