Fabrication of Novel Electrochemical Sensors Based on Modification With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanism and Process of Methylene Blue Degradation by Manganese Oxides Under Microwave Irradiation
Applied Catalysis B: Environmental 160–161 (2014) 211–216 Contents lists available at ScienceDirect Applied Catalysis B: Environmental journal homepage: www.elsevier.com/locate/apcatb Mechanism and process of methylene blue degradation by manganese oxides under microwave irradiation Xiaoyu Wang a, Lefu Mei a, Xuebing Xing a, Libing Liao a,∗, Guocheng Lv a,∗, Zhaohui Li a,b, Limei Wu a a School of Material Sciences and Technology, China University of Geosciences, Beijing 100083, China b Geosciences Department, University of Wisconsin – Parkside, Kenosha, WI 53141-2000, USA article info abstract Article history: Numerous studies have been conducted on the removal of dyes from wastewater, although fewer were Received 21 February 2014 focused on the reactions under the influence of microwave irradiation (MI). In this paper, degradation of Received in revised form 29 April 2014 methylene blue (MB) in simulated wastewater by manganese oxides (MOs) under MI was investigated. A Accepted 5 May 2014 significant increase in MB removal efficiency by akhtenskite or birnessite was observed in the presence Available online 22 May 2014 of MI. After 30 min MI, the MB removal by birnessite was 230 mg/g, in comparison to only 27 mg/g in the absence of MI. The kinetics of MB removal by MOs under MI followed the pseudo-first-order kinetic model Keywords: well with rate constants of 0.005 and 0.04 min−1 for akhtenskite and birnessite, respectively. In contrast, Microwave irradiation −1 Manganese oxides the rate constants were only 0.0006 and 0.0007 min for MB removal by akhtenskite and birnessite in Catalytic oxidation the absence of MI. -

In-Situ Mno2 Electrodeposition and Its Negative Impact to Rechargeable Zinc Manganese Dioxide Batteries
In-situ MnO2 Electrodeposition and its Negative Impact to Rechargeable Zinc Manganese Dioxide Batteries by Rui Lin Liang A thesis presented to the University Of Waterloo in fulfilment of the thesis requirements for the degree of Master of Applied Science in Chemical Engineering (Nanotechnology) Waterloo, Ontario, Canada 2018 © Rui Lin Liang 2018 Author’s Declaration I hereby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final revisions, as accepted by my examiners. I understand that my thesis may be made electronically available to the public. ii Abstract Achieving high rechargeability with the economically feasible and environmentally friendly Zn-MnO2 batteries has been the goal of many scientists in the past half century. Recently, the stability of the system saw a significant improvement through adaptation of mildly acidic electrolyte with Mn2+ additives that prevent dissolution of the active cathode materials, MnO2. With the new design strategy, breakthroughs were made with the battery life span, as the lab scale batteries operated with minimal degradation for over a thousand cycles of charge and discharge at high C- rate ( >5C ) cycling. However, low C-rate operation of these batteries is still limited to 100 cycles, but has not been a focal point of the research efforts. Furthermore, the electrochemical reactions within the battery system are still under debate and many questions remain to be answered. An interesting phenomenon investigated in this thesis about the mildly acidic Zn- MnO2 battery systems is their tendency to experience capacity growth caused by formation of new active material through Mn2+ electrodeposition. -

A Generic Approach for the Synthesis of Nanocrystalline Mesoporous
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 10-10-2014 A Generic Approach for the Synthesis of Nanocrystalline Mesoporous Materials by Inverse Micelle Templating Altug Suleyman Poyraz University of Connecticut - Storrs, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Poyraz, Altug Suleyman, "A Generic Approach for the Synthesis of Nanocrystalline Mesoporous Materials by Inverse Micelle Templating" (2014). Doctoral Dissertations. 569. https://opencommons.uconn.edu/dissertations/569 A Generic Approach for the Synthesis of Nanocrystalline Mesoporous Materials by Inverse Micelle Templating Altug S. Poyraz, PhD University of Connecticut, 2014 There are 4 chapters in this thesis. Chapter 1 provides background information (synthesis, applications, and limitations) about mesoporous materials. Chapter 2 describes the developed inverse micelle method for the synthesis of mesoporous materials and illustrates the applicability of the method. Chapter 3 discusses mesoporous solid acids prepared by inverse micelle method and their catalytic activity. Chapter 4 suggests a mild transformation of mesoporous manganese oxides into various other crystal structures under mild acidic conditions. Thermally stable, crystalline wall, thermally controlled monomodal pore size mesoporous materials are discussed in the thesis. Generation of such materials involves use of inverse micelles, elimination of solvent effects, minimization the effect of water content, and controlling the condensation of inorganic framework by NOx decomposition. Nano-size particles are formed in inverse micelles and are randomly packed to a mesoporous structure. The mesopores are created by interconnected intra-particle voids, thus can be tuned from 1.2 nm to 25 nm by controlling the nano-particle size. -

ABSTRACT BOYD, SHELBY KATHERINE. Interlayer Chemistry
ABSTRACT BOYD, SHELBY KATHERINE. Interlayer Chemistry and Energy Storage Mechanisms of Manganese-Rich Oxides in Aqueous Electrolytes. (Under the direction of Dr. Veronica Augustyn). Electrochemical alkali metal cation insertion from aqueous electrolytes into transition metal (TM) oxides is appealing for low-cost and safe electrochemical energy storage (EES), desalination, and element recovery. The strong interactions between water, electrolyte salts, and TM oxide surfaces dictate the material stability and EES mechanisms. However, these are not yet understood well enough to enable large scale long-life neutral-pH aqueous EES. To advance this understanding I synthesized micron-sized and nanostructured layered manganese-rich oxides and determined how their interlayer environment effects their structural and electrochemical behavior in aqueous electrolytes. First, by tuning the TM content. Second, by tuning the amount of interlayer water. And third, by determining the mechanisms of pseudocapacitive behavior in nanostructured birnessite MnO2. The effect of TM composition on P2 layered Na+ manganese-rich oxides has been extensively investigated for non-aqueous electrolytes, but not aqueous electrolytes. In Chapter 2, I synthesize the following series of P2 oxides and characterize their structural stability upon aqueous electrochemistry: Na0.62Ni0.22Mn0.66Fe0.10O2, Na0.61Ni0.22Mn0.66Co0.10O2, Na0.64Ni0.22Mn0.66Cu0.11O2, and Na0.64Mn0.62Cu0.31O2. Electrochemistry and ex situ X-ray diffraction (XRD) show that water intercalation upon interlayer Na+ removal causes an irreversible phase transformation in all compositions, although transformation extent depends on TM composition and the maximum anodic potential. The 25% c-axis expansion causes eventual electrode failure due to loss of electronic connectivity and particle delamination. -

The Picking Table Volume 50, No. 1
JOURNAL OF THE FRANKLIN-OGDENSBURG MINERALOGICAL SOCIETY Volume 50, No. 1– Spring 2009 $20.00 U.S. Inside This Issue: • Opal added to the Franklin-Sterling Hill species list • Uraninite - a “First Find” from the Trotter Dump • A useful analog to the Franklin-Sterling Hill mineral deposit • Geology of the Hamburg Quarry • A rare mineral discovered in the New Jersey Highlands • How to Photoshop® edit your mineral photos The contents of The Picking Table are licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. The Franklin-Ogdensburg Mineralogical Society, Inc. OFFICERS and STAFF 2009 PRESIDENT (2009-2010) SLIDE COLLECTION CUSTODIAN Bill Truran Edward H. Wilk 2 Little Tarn Court, Hamburg, NJ 07419 202 Boiling Springs Avenue (973) 827-7804 E. Rutherford, NJ 07073 [email protected] (201) 438-8471 VICE-PRESIDENT (2009-2010) TRUSTEES Richard Keller C. Richard Bieling (2009-2010) 13 Green Street, Franklin, NJ 07416 Richard C. Bostwick (2009-2010) (973) 209-4178 George Elling (2008-2009) [email protected] Steven M. Kuitems (2009-2010) Chester S. Lemanski, Jr. (2008-2009) SECOND VICE-PRESIDENT (2009-2010) Lee Lowell (2008-2009) Joe Kaiser Earl Verbeek (2008-2009) 40 Castlewood Trail, Sparta, NJ 07871 Edward H. Wilk (2008-2009) (973) 729-0215 Fred Young (2008-2009) [email protected] LIAISON WITH THE EASTERN FEDERATION SECRETARY OF MINERALOGICAL AND LAPIDARY Tema J. Hecht SOCIETIES (EFMLS) 600 West 111TH Street, Apt. 11B Delegate Joe Kaiser New York, NY 10025 Alternate Richard C. Bostwick (212) 749-5817 (Home) (917) 903-4687 (Cell) COMMITTEE CHAIRPERSONS [email protected] Auditing William J. Trost Field Trip Warren Cummings TREASURER Historical John L. -

Supplementary Information Mitigating Effect of Organic Matter on the in Vivo Toxicity of Metal Oxide Nanoparticles in the Marine Environment
Electronic Supplementary Material (ESI) for Environmental Science: Nano. This journal is © The Royal Society of Chemistry 2018 Supplementary Information Mitigating effect of organic matter on the in vivo toxicity of metal oxide nanoparticles in the marine environment Seta Noventa*a, Darren Rowea and Tamara Gallowaya *corresponding author: email: [email protected], tel: 01392 263436 a College of Life and Environmental Sciences, University of Exeter, EX4 4QD, Exeter UK, This file includes additional information about: Primary physico-chemical properties of the test-NPs and methods used for the characterization Methods for the characterization of the behaviour of the test-NPs Primer sequences, qPCR conditions and performance MnO2 NP aggregation kinetics MnO2 NP ingestion TEM-EDS study of cellular internalization of MnO2 NPs TEM-EDS analyses of electron dense particles suspected of being cellular internalized BSA coated MnO2 NPs Abbreviations: NPs: nanoparticles; TEM: Transmission Electron Microscopy; BET: Branauer, Emmett and Teller; SEM: Scanning Electron Microscope; EDS: Energy Dispersive X-ray Spectroscopy; pHIEP: pH at the isoelectric point; DIW: deionised water; ASW: artificial seawater; Cyt c: Cytochrome c. Primary physico-chemical properties of the test-NPs and methods used for the characterization The primary physico-chemical proprieties of the test NPs, ZnO NPs, and MnO2 NPs, are summarized in Table S1. Data was retrieved from official reports (for ZnO NPs only 1) and supplemented with data performed at the NERC Facility for Environmental Nanoscience Analysis and Characterisation (FENAC, Birmingham, UK). The description of the methods used and more details about the results obtained are reported below. Table S1. Primary physico-chemical proprieties of the test NPs, ZnO NPs and MnO2 NPs. -

Mineral Names, Redefinitions & Discreditations Passed by the CNMMN of The
9 February 2004 – Note to our readers: We welcome your comments and criticism. If you are reporting substantive changes, errors or omissions, please provide us with a literature reference so we can confirm what you have suggested. Thanks! Mineral Names, Redefinitions & Discreditations Passed by the CNMMN of the IMA Ernie Nickel & Monte Nichols [email protected] & [email protected] Aleph Enterprises PO Box 213 Livermore, California 94551 USA This list contains minerals derived from the Materials Data, Inc. MINERAL Database and was produced expressly for the use of the Commission on New Minerals and Mineral Names (CNMMN) of the International Mineralogical Association (IMA). All rights to the use of the data presented here, either printed or electronic, rest with Materials Data, Inc. Minerals presented include those voted as “approved” (A), “redefined or renamed” (R) and “discredited” (D) species by the CNMMN as well as a number of former mineral names the CNMMN decided would better be used as group names (g). Their abbreviated symbol appears at the left margin. Minerals in the MINERAL Database which have been designated as “Q” (questionable minerals, not approved by the CNMMN), as “G” (“golden or grandfathered” minerals described in the past and generally believed to represent valid species names), as “U” (unnamed), as “N” (not approved by the CNMMN) and as “P” (polymorphs) have not been included as part of this listing. These minerals can be found in the Free MINERAL Database now being distributed by Materials Data, Inc ([email protected] and http://www.materialsdata.com) for just the cost of making and shipping the CD or without any cost whatever when downloaded directly from the Materials Data website. -

Supporting Information
ORE Open Research Exeter TITLE Dissolution and bandgap paradigms for predicting the toxicity of metal oxide nanoparticles in the marine environment: an in vivo study with oyster embryos AUTHORS Noventa, S; Hacker, C; Rowe, D; et al. JOURNAL Nanotoxicology DEPOSITED IN ORE 29 August 2018 This version available at http://hdl.handle.net/10871/33846 COPYRIGHT AND REUSE Open Research Exeter makes this work available in accordance with publisher policies. A NOTE ON VERSIONS The version presented here may differ from the published version. If citing, you are advised to consult the published version for pagination, volume/issue and date of publication Supporting Information Dissolution and Bandgap paradigms for predicting the toxicity of metal oxide nanoparticles in the marine environment: an in vivo study with oyster embryos Seta Noventa*a, Christian Hackerb, Darren Rowea, Christine Elgyc and Tamara Gallowaya *corresponding author: email: [email protected], tel: 01392 263436 a College of Life and Environmental Sciences, University of Exeter, EX4 4QD, Exeter UK, b College of Life and Environmental Sciences, Bioimaging Centre, University of Exeter, EX4 4QD, Exeter UK c Department of Geography, Earth and Environmental Sciences, Facility for Environmental Nanoscience Analysis and Characterisation, University of Birmingham, Edgbaston, Birmingham, UK This file includes additional information about: • Primary physico-chemical properties of the test-NPs • Primer sequences, qPCR conditions and performance • TEM-EDS study of biological fate of MnO2 NPs • Methods for the characterization of the behaviour of the test-NPs • Results of the Cytochrome c oxidation assay carried out on ZnO and CeO2 NP dispersions in deionized water Abbreviations: NPs: nanoparticles; TEM: Transmission Electron Microscopy; BET: Branauer, Emmett and Teller; EELS: Electron Energy Loss Spectroscopy; SEM: Scanning Electron Microscope; EDS: Energy Dispersive X-ray Spectroscopy; DIW: deionized water; ASW: artificial seawater; Cyt c: Cytochrome c; pHIEP: pH at the isoelectric point. -
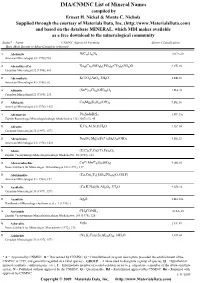
IMA/CNMNC List of Mineral Names
IMA/CNMNC List of Mineral Name s compiled by Ernest H. Nickel & Monte C. Nichols Supplied through the courtesy of Materials Data, Inc. (http://www.MaterialsData.com) and based on the database MINERAL, which MDI makes available as a free download to the mineralogical community Status* Name CNMNC Approved Formula Strunz Classification Best, Most Recent or Most Complete reference. A Abelsonite NiC£¡H£¢N¤ 10.CA.20 American Mineralogist 63 (1978) 930 A Abenakiite-(Ce) Na¢¦Ce¦(SiO£)¦(PO¤)¦(CO£)¦(SO¢)O 9.CK.10 Canadian Mineralogist 32 (1994), 843 G Abernathyite K(UO¢)AsO¤•3H¢O 8.EB.15 American Mineralogist 41 (1956), 82 A Abhurite (SnÀÈ)¢¡Cl¡¦(OH)¡¤O¦ 3.DA.30 Canadian Mineralogist 23 (1985), 233 D Abkhazite Ca¢Mg¥Si¨O¢¢(OH)¢ 9.DE.10 American Mineralogist 63 (1978), 1023 A Abramovite Pb¢SnInBiS§ 2.HF.25a Zapiski Rossiiskogo Mineralogicheskogo Obshchetstva 136 (2007) (5), 45 D Abrazite K,Ca,Al,Si,O,H¢O 9.GC.05 Canadian Mineralogist 35 (1997), 1571 D Abriachanite Na¢(Fe,Mg)£(FeÁÈ)¢Si¨O¢¢(OH)¢ 9.DE.25 American Mineralogist 63 (1978), 1023 D Absite (U,Ca,Y,Ce)(Ti,Fe)¢O¦ Zapiski Vsesoyuznogo Mineralogicheskogo Obshchestva 92 (1963), 113 A Abswurmbachite CuÀÈ(MnÁÈ)¦O¨(SiO¤) 9.AG.05 Neues Jahrbuch für Mineralogie, Abhandlungen 163 (1991), 117 D Abukumalite (Ca,Ce)¢Y£(SiO¤,PO¤)£(O,OH,F) American Mineralogist 51 (1966), 152 D Acadialite (Ca,K,Na)(Si,Al)£O¦•3H¢O 9.GD.10 Canadian Mineralogist 35 (1997), 1571 G Acanthite Ag¢S 2.BA.30a Handbook of Mineralogy (Anthony et al.), 1 (1990), 1 A Acetamide CH£CONH¢ 10.AA.20 Zapiski Vsesoyuznogo Mineralogicheskogo -
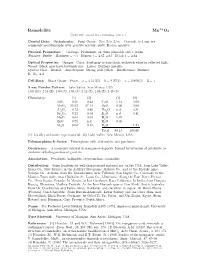
Ramsdellite Mn O2 C 2001-2005 Mineral Data Publishing, Version 1
4+ Ramsdellite Mn O2 c 2001-2005 Mineral Data Publishing, version 1 Crystal Data: Orthorhombic. Point Group: 2/m 2/m 2/m. Crystals, to 1 cm, are commonly pseudomorphs after groutite crystals; platy, fibrous, massive. Physical Properties: Cleavage: Prominent, on three pinacoids and a prism. Tenacity: Brittle. Hardness = ∼3 D(meas.) = 4.65–4.83 D(calc.) = 4.84 Optical Properties: Opaque. Color: Steel-gray to iron-black; yellowish white in reflected light. Streak: Black, may have brownish tint. Luster: Brilliant metallic. Optical Class: Biaxial. Anisotropism: Strong; pale yellow. Bireflectance: Distinct. R1–R2: n.d. Cell Data: Space Group: P bnm. a = 4.533(5) b = 9.27(1) c = 2.866(5) Z = 4 X-ray Powder Pattern: Lake Valley, New Mexico, USA. 4.08 (10), 2.53 (8), 1.60 (7), 1.64 (6), 2.13 (5), 1.88 (5), 1.46 (5) Chemistry: (1) (2) (1) (2) SiO2 0.36 0.42 CaO 1.12 0.00 MnO2 95.02 97.14 BaO 0.00 0.00 Al2O3 0.72 0.48 Na2O n.d. 0.31 Fe2O3 0.22 0.34 K2O n.d. 0.41 + MnO 0.00 0.00 H2O 1.39 − ZnO 0.25 n.d. H2O 0.05 MgO 0.00 0.12 H2O 1.24 Total 99.13 100.46 (1) Locality unknown; type material. (2) Lake Valley, New Mexico, USA. Polymorphism & Series: Trimorphous with akhtenskite and pyrolusite. Occurrence: A secondary mineral in manganese deposits, formed by inversion of pyrolusite or oxidative dehydrogenation of groutite. Association: Pyrolusite, hollandite, cryptomelane, coronadite. -

Akhtenskite Mno2 C 2001-2005 Mineral Data Publishing, Version 1
Akhtenskite MnO2 c 2001-2005 Mineral Data Publishing, version 1 Crystal Data: Hexagonal. Point Group: 6/m 2/m 2/m. As microscopic crystals, flaky to platy on {001}, in parallel aggregates, sometimes in rows at 120◦, probably due to replacement of an earlier hexagonal mineral; as flaky polycrystalline aggregates. Physical Properties: Cleavage: {001}. Hardness = n.d. D(meas.) = n.d. D(calc.) = [4.78] Optical Properties: [Opaque.] Color: Pale gray to black. Optical Class: Uniaxial. R1–R2: n.d. Cell Data: Space Group: P 63/mmc. a = 2.83–2.85 c = 4.47–4.88 Z = [1] X-ray Powder Pattern: Mt. Zarod, Russia; calculated from an electron diffraction pattern. 2.45, 2.15, 1.65, 1.42 Chemistry: Sufficient material for direct chemical analysis cannot be separated; energy-dispersive analysis shows Mn as the only cationic species; Mn4+ and O were established by X-ray photoelectronic spectroscopy, as was the absence of OH and H2O. Polymorphism & Series: Trimorphous with pyrolusite and ramsdellite. Occurrence: In mixtures in “psilomelane” with other manganese oxides in an iron oxide deposit, probably bacterially altered from a previous mineral (Akhtensk deposit, Russia); in incrustations of ferromanganese minerals on oceanic basalt on a guyot (Mt. Zarod, Russia). Association: Cryptomelane, nsutite, pyrolusite, todorokite, goethite (Akhtensk deposit, Russia); vernadite, manganite, Fe–Mn oxides (Mt. Zarod, Russia). Distribution: In the Akhtensk brown ironstone deposit, north of Magnitka, Southern Ural Mountains; on Mt. Zarod, Sikhote-Alin Mountains, Primorskiy Kray, Russia. Name: For the Akhtensk deposit, Russia, where it was first noted. Type Material: Mining Institute, St. Petersburg, Russia, 307/5. -

Erroneous Attribution of Mn-Oxides Minerals in Primary Mineralogy
MUSEOLOGIA SCIENTIFICA nuova serie • 13: 96-102 • 2019 ISSN 1123-265X Professionalità / Gestione Erroneous attribution of Mn-oxides minerals in primary mineralogy collections Emanuele Costa - Silvia Ronchetti - Annalau- Emanuele Costa ra Pistarino - Alessandro Delmastro - Lorenzo Dipartimento di Scienze della Terra, Università degli Studi di Torino, Via Valperga Caluso, 35. I-10125 Torino. Mariano Gallo - Massimo Umberto Tomalino E-mail: [email protected] (corresponding author) Silvia Ronchetti Dipartimento Scienza Applicata e Tecnologia (DISAT), Politecnico di Torino, Corso Duca degli Abruzzi, 24. I-10129 Torino. Annalaura Pistarino Museo Regionale di Scienze Naturali, Via Giovanni Giolitti, 36. I-10123 Torino. Alessandro Delmastro Dipartimento di Ingegneria dell’Ambiente, del Territorio e delle Infrastrutture (DIATI), Politecnico di Torino, Corso Duca degli Abruzzi, 24. I-10129 Torino. Lorenzo Mariano Gallo Former Museo Regionale di Scienze Naturali, Via Giovanni Giolitti, 36. I-10123 Torino. Massimo Umberto Tomalino Dipartimento Scienza Applicata e Tecnologia (DISAT), Politecnico di Torino, Corso Duca degli Abruzzi, 24. I-10129 Torino. ABSTRACT The manganese oxides, hydro-oxides and closely related species form a group of minerals mainly resembling black or dark gray powder, black mammillons, black tiny crystals, black thin deposits within rock fissures and along veins. The distinction of the different species, based on the external appearance, is still difficult, although not as much as in the past, when knowledge and practice on the instrumental techniques were not available. In spite of the interest in the museum and mineral collecting community, detailed qualitative analyses and structural characterization have rarely been carried out on museum samples. In this study, we focus on the identification of a significant number of specimens from historic and modern collections.