61: Calcium Channel Blockers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pharmacology
Pharmacology New for 2020-2021 Competitor orientation deleted from ILC. Event Summary Pharmacology provides HOSA members with the opportunity to gain knowledge and skills regarding the area of healthcare concerned with uses, effects, and modes of actions of drugs. This competitive event consists of a written test with a tie-breaker essay question. This event aims to inspire members to learn about how drugs work in the body, proper administration, and adaptations for different patients and conditions. Dress Code Competitors must be in official HOSA uniform or proper business attire. Bonus points will be awarded for proper dress. General Rules 1. Competitors in this event must be active members of HOSA-Future Health Professionals, in good standing. 2. Secondary and Postsecondary/Collegiate divisions are eligible to compete in this event. 3. Competitors must be familiar with and adhere to the “General Rules and Regulations of the HOSA Competitive Events Program (GRR)." 4. All competitors shall report to the site of the event at the time designated for each round of competition. At ILC, competitor’s photo ID must be presented prior to ALL competition rounds. Official References • Fulcher, Soto and Fulcher. Pharmacology: Principles and Applications. Elsevier, Latest edition. • Ford, Susan and Sally Roach. Roach’s Introductory Clinical Pharmacology. Wolters Kluwer, Latest edition. Written Test 5. Test Instructions: The written test will consist of 100 multiple choice items in a maximum of 90 minutes. 6. Time Remaining Announcements: There will be a verbal announcement when there are 60 minutes, 30 minutes, 15 minutes, 5 minutes, and 1 minute remaining to complete the test. -

Selective Mtorc2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival
Author Manuscript Published OnlineFirst on January 22, 2018; DOI: 10.1158/0008-5472.CAN-17-2388 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Selective mTORC2 inhibitor therapeutically blocks breast cancer cell growth and survival Thomas A. Werfel1, 3, Shan Wang2, Meredith A. Jackson1, Taylor E. Kavanaugh1, Meghan Morrison Joly3, Linus H. Lee1, Donna J. Hicks3, Violeta Sanchez4, Paula Gonzalez Ericsson4, Kameron V. Kilchrist1, Somtochukwu C. Dimobi1, Samantha M. Sarett1, Dana Brantley-Sieders2, Rebecca S. Cook1,3,4* and Craig L. Duvall1* 1Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37232 USA 2Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232.USA 3Department of Cell and Developmental Biology, Vanderbilt University School of Medicine, Nashville, TN 37232 USA 4Breast Cancer Research Program, Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, TN 37232 USA Running Title: A selective mTORC2 inhibitor blocks breast cancer growth Key Words: Breast Cancer, mTOR, Rictor, RNA interference, Nanomedicine *To whom correspondence should be addressed: Craig L. Duvall, PhD Vanderbilt University School of Engineering Department of Biomedical Engineering Nashville, TN 37232 Phone: (615) 322-3598 Fax: (615) 343-7919 Email: [email protected] Rebecca S. Cook, PhD Vanderbilt University Medical Center Department of Cancer Biology Nashville, TN 37232 Phone: (615) 936-3813 Fax: (615) 936-3811 Email: [email protected] Funding. This work was supported by Specialized Program of Research Excellence (SPORE) grant NIH P50 CA098131 (VICC), Cancer Center Support grant NIH P30 CA68485 (VICC), NIH F31 CA195989-01 (MMW), NIH R01 EB019409, DOD CDMRP OR130302, NSF GFRP 1445197, and CTSA UL1TR000445 from the National Center for Advancing Translational Sciences. -

ADHD Parents Medication Guide Revised July 2013
ADHD Parents Medication Guide Revised July 2013 Attention-Deficit/Hyperactivity Disorder Prepared by: American Academy of Child & Adolescent Psychiatry and American Psychiatric Association Supported by the Elaine Schlosser Lewis Fund Physician: ___________________________________________________ Address: ___________________________________________________ ___________________________________________________ ___________________________________________________ Phone: ___________________________________________________ Email: ___________________________________________________ ADHD Parents Medication Guide – July 2013 2 Introduction Attention-Deficit/Hyperactivity Disorder (ADHD) is a neurodevelopmental disorder characterized by difficulty paying attention, excessive activity, and impulsivity (acting before you think). ADHD is usually identified when children are in grade school but can be diagnosed at any time from preschool to adulthood. Recent studies indicate that almost 10 percent of children between the ages of 4 to 17 are reported by their parents as being diagnosed with ADHD. So in a classroom of 30 children, two to three children may have ADHD.1,2,3,4,5 Short attention spans and high levels of activity are a normal part of childhood. For children with ADHD, these behaviors are excessive, inappropriate for their age, and interfere with daily functioning at home, school, and with peers. Some children with ADHD only have problems with attention; other children only have issues with hyperactivity and impulsivity; most children with ADHD have problems with all three. As they grow into adolescence and young adulthood, children with ADHD may become less hyperactive yet continue to have significant problems with distraction, disorganization, and poor impulse control. ADHD can interfere with a child’s ability to perform in school, do homework, follow rules, and develop and maintain peer relationships. When children become adolescents, ADHD can increase their risk of dropping out of school or having disciplinary problems. -
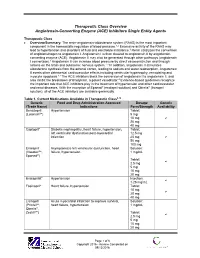
Angiotensin-Converting Enzyme (ACE) Inhibitors Single Entity Agents
Therapeutic Class Overview Angiotensin-Converting Enzyme (ACE) Inhibitors Single Entity Agents Therapeutic Class Overview/Summary: The renin-angiotensin-aldosterone system (RAAS) is the most important component in the homeostatic regulation of blood pressure.1,2 Excessive activity of the RAAS may lead to hypertension and disorders of fluid and electrolyte imbalance.3 Renin catalyzes the conversion of angiotensinogen to angiotensin I. Angiotensin I is then cleaved to angiotensin II by angiotensin- converting enzyme (ACE). Angiotensin II may also be generated through other pathways (angiotensin I convertase).1 Angiotensin II can increase blood pressure by direct vasoconstriction and through actions on the brain and autonomic nervous system.1,3 In addition, angiotensin II stimulates aldosterone synthesis from the adrenal cortex, leading to sodium and water reabsorption. Angiotensin II exerts other detrimental cardiovascular effects including ventricular hypertrophy, remodeling and myocyte apoptosis.1,2 The ACE inhibitors block the conversion of angiotensin I to angiotensin II, and also inhibit the breakdown of bradykinin, a potent vasodilator.4 Evidence-based guidelines recognize the important role that ACE inhibitors play in the treatment of hypertension and other cardiovascular and renal diseases. With the exception of Epaned® (enalapril solution) and Qbrelis® (lisinopril solution), all of the ACE inhibitors are available generically. Table 1. Current Medications Available in Therapeutic Class5-19 Generic Food and Drug Administration -

Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development
Clinical Pharmacology 1: Phase 1 Studies and Early Drug Development Gerlie Gieser, Ph.D. Office of Clinical Pharmacology, Div. IV Objectives • Outline the Phase 1 studies conducted to characterize the Clinical Pharmacology of a drug; describe important design elements of and the information gained from these studies. • List the Clinical Pharmacology characteristics of an Ideal Drug • Describe how the Clinical Pharmacology information from Phase 1 can help design Phase 2/3 trials • Discuss the timing of Clinical Pharmacology studies during drug development, and provide examples of how the information generated could impact the overall clinical development plan and product labeling. Phase 1 of Drug Development CLINICAL DEVELOPMENT RESEARCH PRE POST AND CLINICAL APPROVAL 1 DISCOVERY DEVELOPMENT 2 3 PHASE e e e s s s a a a h h h P P P Clinical Pharmacology Studies Initial IND (first in human) NDA/BLA SUBMISSION Phase 1 – studies designed mainly to investigate the safety/tolerability (if possible, identify MTD), pharmacokinetics and pharmacodynamics of an investigational drug in humans Clinical Pharmacology • Study of the Pharmacokinetics (PK) and Pharmacodynamics (PD) of the drug in humans – PK: what the body does to the drug (Absorption, Distribution, Metabolism, Excretion) – PD: what the drug does to the body • PK and PD profiles of the drug are influenced by physicochemical properties of the drug, product/formulation, administration route, patient’s intrinsic and extrinsic factors (e.g., organ dysfunction, diseases, concomitant medications, -

I. Antihistamines Seunghoon Han* Department of Clinical Pharmacology and Therapeutics, Seoul St
2014;22(1):13-18 TCP Translational and Clinical Pharmacology http://dx.doi.org/10.12793/tcp.2014.22.1.13 Clinical Pharmacology Review for Primary Health Care Providers: I. Antihistamines Seunghoon Han* Department of Clinical Pharmacology and Therapeutics, Seoul St. Mary’s Hospital, The Catholic University of Korea, Seoul 137-701, Korea *Correspondence: S. Han; Tel: +82-2-2258-7326, Fax: +82-2-2258-7876, E-mail: [email protected] Received 31 May 2014 Primary health care providers play a critical role in maintaining public health, and the appropri- Accepted 31 May 2014 ate use of pharmaceutical products is one of the major parts of their practice. This series of articles, pISSN: 2289-0882 entitled ‘Clinical Pharmacology Review for Primary Health Care Providers,’ is intended to help pri- mary health care providers select more appropriate prescriptions for frequently used drugs based on up-to-date information. We expect that this effort will contribute to improvements in public health and diminish unnecessary drug use. Introduction tion on the H1 receptor.[8] THERAPEU Antihistamines include some of the most frequently prescribed drugs in the primary health care environment for the symp- Generations and Classes tomatic relief of allergic diseases, the common cold, urticaria, Many primary health care providers are well-informed about T and insomnia.[1-5] The importance of antihistamines has been the different ‘generations’ of antihistamines but not about the ICS TU emphasized as the prevalence of target diseases increases.[6,7] different ‘classes’ characterized according to chemical structure. However, the appropriate use, clinical effectiveness, and target [1] This discrepancy seems reasonable because ‘inter-generation’ T populations for prescription of antihistamines are still a matter differences are more prominent than ‘inter-class’ differences. -

Guidance for Reviewers Pharmacology/Toxicology Review Format
Guidance for Reviewers Pharmacology/Toxicology Review Format U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Pharmacology/Toxicology May 2001 Guidance for Reviewers Pharmacology/Toxicology Review Format Additional copies are available from: Drug Information Branch (HFD-210) Center for Drug Evaluation and Research (CDER) 5600 Fishers Lane, Rockville, MD 20857, (Tel) 301-827-4570 Internet at http://www.fda.gov/der/guidance/index.htm U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Pharmacology/Toxicology May 2001 Guidance for Reviewers Table of Contents I. INTRODUCTION ....................................................................................................1 II. BACKGROUND.......................................................................................................1 III. DISCUSSION ...........................................................................................................1 A. IND REVIEW FORMAT ....................................................................................1 B. NDA REVIEW FORMAT...................................................................................1 IV. ATTACHMENTS .....................................................................................................1 A. IND REVIEW FORMAT ....................................................................................A-i B. NDA REVIEW FORMAT...................................................................................B-i -

Department of Pharmacology (GRAD) 1
Department of Pharmacology (GRAD) 1 faculty participate fully at all levels. The department has the highest DEPARTMENT OF level of NIH funding of all pharmacology departments and a great diversity of research areas is available to trainees. These areas PHARMACOLOGY (GRAD) include cell surface receptors, G proteins, protein kinases, and signal transduction mechanisms; neuropharmacology; nucleic acids, cancer, Contact Information and antimicrobial pharmacology; and experimental therapeutics. Cell and molecular approaches are particularly strong, but systems-level research Department of Pharmacology such as behavioral pharmacology and analysis of knock-in and knock-out Visit Program Website (http://www.med.unc.edu/pharm/) mice is also well-represented. Excellent physical facilities are available for all research areas. Henrik Dohlman, Chair Students completing the training program will have acquired basic The Department of Pharmacology offers a program of study that leads knowledge of pharmacology and related fields, in-depth knowledge in to the degree of doctor of philosophy in pharmacology. The curriculum is their dissertation research area, the ability to evaluate scientific literature, individualized in recognition of the diverse backgrounds and interests of mastery of a variety of laboratory procedures, skill in planning and students and the broad scope of the discipline of pharmacology. executing an important research project in pharmacology, and the ability The department offers a variety of research areas including to communicate results, analysis, and interpretation. These skills provide a sound basis for successful scientific careers in academia, government, 1. Receptors and signal transduction or industry. 2. Ion channels To apply to BBSP, students must use The Graduate School's online 3. -

Beta Blocker
Medication Information Beta Blocker Other names for this medication Acebutol Nadolol Atenolol Pindolol Bisoprolol Propanolol Carvediol Sotalol Labetalol Timolol Metoprolol There are many other names for this medication. How this medication is used This medication causes your heart to beat slower. This helps rest the heart after a heart attack. This medication helps prevent and/or reduce chest pain and irregular heart beats. It does not stop chest pain or angina after the pain has started. Bisoprolol, Carvedilol and Metoprolol can be used to prevent heart failure. They work by relaxing the blood vessels. This allows more blood to go to the heart. The more blood that goes to the heart, the more oxygen the heart gets. This helps the heart work better. Some of these medications are used to treat high blood pressure, migraine headaches and muscle tremors. Beta Blocker How to take this medication Take this medication exactly as directed by your doctor or health care provider. It must be taken regularly, even if you feel well. Do not suddenly stop taking this medication without checking with your doctor or health care provider first. Suddenly stopping this medication can cause: • chest pain • irregular heart beats • high blood pressure. When it is time to stop taking this medication your doctor or health care provider may slowly decrease the amount. If you miss a dose of this medication, take it as soon as possible. However, if it is almost time for the next dose, skip the missed dose and go back to your regular time. Do not take 2 doses at one time. -

Psychiatric Medications in Behavioral Healthcarev5
Copyright © The University of South Florida, 2012 Psychiatric Medications in Behavioral Healthcare An Important Notice None of the pages in this tutorial are meant to be a replacement for professional help. The science of medicine is constantly changing, and these changes alter treatment and drug therapies as a result of what is learned through research and clinical experience. The author has relied on resources believed to be reliable at the time material was developed. However, there is always the possibility of human error or changes in medical science and neither the authors nor the University of South Florida can guarantee that all the information in this program is in every respect accurate or complete and they are not responsible for any errors or omissions or for the results obtained from the use of such information. Each person that reads this program is encouraged to confirm the information with other sources and understand that it not be interpreted as medical or professional advice. All medical information needs to be carefully reviewed with a health care provider. Course Objectives At the completion of this program participants should be able to: • Identify at 5 categories of medications used to treat the symptoms of psychiatric disorders, the therapeutic effects of medications in each category, and the side effects associated with medications in each category. • Identify at least 5 medications and the benefits of those medications as compared to the medications. • Identify at least 5 reasons that a person may stop taking medications or not take medications as prescribed. • Demonstrate your learned understanding of psychiatric medications by passing the combined post‐ tests. -

The Psychoactive Effects of Psychiatric Medication: the Elephant in the Room
Journal of Psychoactive Drugs, 45 (5), 409–415, 2013 Published with license by Taylor & Francis ISSN: 0279-1072 print / 2159-9777 online DOI: 10.1080/02791072.2013.845328 The Psychoactive Effects of Psychiatric Medication: The Elephant in the Room Joanna Moncrieff, M.B.B.S.a; David Cohenb & Sally Porterc Abstract —The psychoactive effects of psychiatric medications have been obscured by the presump- tion that these medications have disease-specific actions. Exploiting the parallels with the psychoactive effects and uses of recreational substances helps to highlight the psychoactive properties of psychi- atric medications and their impact on people with psychiatric problems. We discuss how psychoactive effects produced by different drugs prescribed in psychiatric practice might modify various disturb- ing and distressing symptoms, and we also consider the costs of these psychoactive effects on the mental well-being of the user. We examine the issue of dependence, and the need for support for peo- ple wishing to withdraw from psychiatric medication. We consider how the reality of psychoactive effects undermines the idea that psychiatric drugs work by targeting underlying disease processes, since psychoactive effects can themselves directly modify mental and behavioral symptoms and thus affect the results of placebo-controlled trials. These effects and their impact also raise questions about the validity and importance of modern diagnosis systems. Extensive research is needed to clarify the range of acute and longer-term mental, behavioral, and physical effects induced by psychiatric drugs, both during and after consumption and withdrawal, to enable users and prescribers to exploit their psychoactive effects judiciously in a safe and more informed manner. -

Pharmacology 101
Pharmacology 101 Tyler Fischback, PharmD, BCPS, DPLA Clinical Pharmacy Manager Confluence Health Wenatchee, WA Objectives • Define Pharmacology, Pharmacokinetics and Pharmacodynamics • Understand how drug interactions work • Understand how some specific drugs behave in the body (opioids, benzodiazepines, amiodarone) • Apply pharmacology principles into practice Medication Errors and Adverse Drug Events • Error: An error of commission or omission at any step along the pathway that begins when a clinician prescribes a medication and ends when the patient actually received the medication. • ADE: Harm experienced by a patient as a result of exposure to a medication. ADE does not necessarily indicate an error or poor care. However, ~1/2 of ADEs are preventable. Anyone here ever seen a medication error or adverse drug event? Anyone here ever made a medication error? How many different prescription medications are available on the U.S. market? 1,000 So, it’s no surprise why we see so many problems………………… EXCEPT……. The real number is 10,000 Adverse Drug Events • ~1/3 of U.S. adults use 5 or more medications • Annually, ADE = 700,000 ER visits and 100,000 hospitalizations • So, is pharmacology important to your work? • Additionally, 5% of hospitalized patient experience an ADE during their stay • High risk: Anticoagulants, Opioids, Insulin, Cardiac, and Transitions of Care Adverse Drug Events, cont. • Elderly are more susceptible • Pediatrics patients more susceptible (weight-based dosing), especially liquids • Caregivers and patients admittedly