Identification and Characterization of Novel Astroviruses Stacy Finkbeiner Washington University in St
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of an Overprinting Gene in Merkel Cell Polyomavirus Provides Evolutionary Insight Into the Birth of Viral Genes
Identification of an overprinting gene in Merkel cell polyomavirus provides evolutionary insight into the birth of viral genes Joseph J. Cartera,b,1,2, Matthew D. Daughertyc,1, Xiaojie Qia, Anjali Bheda-Malgea,3, Gregory C. Wipfa, Kristin Robinsona, Ann Romana, Harmit S. Malikc,d, and Denise A. Gallowaya,b,2 Divisions of aHuman Biology, bPublic Health Sciences, and cBasic Sciences and dHoward Hughes Medical Institute, Fred Hutchinson Cancer Research Center, Seattle, WA 98109 Edited by Peter M. Howley, Harvard Medical School, Boston, MA, and approved June 17, 2013 (received for review February 24, 2013) Many viruses use overprinting (alternate reading frame utiliza- mammals and birds (7, 8). Polyomaviruses leverage alternative tion) as a means to increase protein diversity in genomes severely splicing of the early region (ER) of the genome to generate pro- constrained by size. However, the evolutionary steps that facili- tein diversity, including the large and small T antigens (LT and ST, tate the de novo generation of a novel protein within an ancestral respectively) and the middle T antigen (MT) of murine poly- ORF have remained poorly characterized. Here, we describe the omavirus (MPyV), which is generated by a novel splicing event and identification of an overprinting gene, expressed from an Alter- overprinting of the second exon of LT. Some polyomaviruses can nate frame of the Large T Open reading frame (ALTO) in the early drive tumorigenicity, and gene products from the ER, especially region of Merkel cell polyomavirus (MCPyV), the causative agent SV40 LT and MPyV MT, have been extraordinarily useful models of most Merkel cell carcinomas. -

A Preliminary Study of Viral Metagenomics of French Bat Species in Contact with Humans: Identification of New Mammalian Viruses
A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses. Laurent Dacheux, Minerva Cervantes-Gonzalez, Ghislaine Guigon, Jean-Michel Thiberge, Mathias Vandenbogaert, Corinne Maufrais, Valérie Caro, Hervé Bourhy To cite this version: Laurent Dacheux, Minerva Cervantes-Gonzalez, Ghislaine Guigon, Jean-Michel Thiberge, Mathias Vandenbogaert, et al.. A preliminary study of viral metagenomics of French bat species in contact with humans: identification of new mammalian viruses.. PLoS ONE, Public Library of Science, 2014, 9 (1), pp.e87194. 10.1371/journal.pone.0087194.s006. pasteur-01430485 HAL Id: pasteur-01430485 https://hal-pasteur.archives-ouvertes.fr/pasteur-01430485 Submitted on 9 Jan 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License A Preliminary Study of Viral Metagenomics of French Bat Species in Contact with Humans: Identification of New Mammalian Viruses Laurent Dacheux1*, Minerva Cervantes-Gonzalez1, -

Guide for Common Viral Diseases of Animals in Louisiana
Sampling and Testing Guide for Common Viral Diseases of Animals in Louisiana Please click on the species of interest: Cattle Deer and Small Ruminants The Louisiana Animal Swine Disease Diagnostic Horses Laboratory Dogs A service unit of the LSU School of Veterinary Medicine Adapted from Murphy, F.A., et al, Veterinary Virology, 3rd ed. Cats Academic Press, 1999. Compiled by Rob Poston Multi-species: Rabiesvirus DCN LADDL Guide for Common Viral Diseases v. B2 1 Cattle Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 2 Deer and Small Ruminants Please click on the principle system involvement Generalized viral disease Respiratory viral disease Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 3 Swine Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 4 Horses Please click on the principle system involvement Generalized viral diseases Neurological viral diseases Respiratory viral diseases Enteric viral diseases Abortifacient/neonatal viral diseases Viral infections affecting the skin Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. B2 5 Dogs Please click on the principle system involvement Generalized viral diseases Respiratory viral diseases Enteric viral diseases Reproductive/neonatal viral diseases Back to the Beginning DCN LADDL Guide for Common Viral Diseases v. -

Key Factors That Enable the Pandemic Potential of RNA Viruses and Inter-Species Transmission: a Systematic Review
viruses Review Key Factors That Enable the Pandemic Potential of RNA Viruses and Inter-Species Transmission: A Systematic Review Santiago Alvarez-Munoz , Nicolas Upegui-Porras , Arlen P. Gomez and Gloria Ramirez-Nieto * Microbiology and Epidemiology Research Group, Facultad de Medicina Veterinaria y de Zootecnia, Universidad Nacional de Colombia, Bogotá 111321, Colombia; [email protected] (S.A.-M.); [email protected] (N.U.-P.); [email protected] (A.P.G.) * Correspondence: [email protected]; Tel.: +57-1-3-16-56-93 Abstract: Viruses play a primary role as etiological agents of pandemics worldwide. Although there has been progress in identifying the molecular features of both viruses and hosts, the extent of the impact these and other factors have that contribute to interspecies transmission and their relationship with the emergence of diseases are poorly understood. The objective of this review was to analyze the factors related to the characteristics inherent to RNA viruses accountable for pandemics in the last 20 years which facilitate infection, promote interspecies jump, and assist in the generation of zoonotic infections with pandemic potential. The search resulted in 48 research articles that met the inclusion criteria. Changes adopted by RNA viruses are influenced by environmental and host-related factors, which define their ability to adapt. Population density, host distribution, migration patterns, and the loss of natural habitats, among others, have been associated as factors in the virus–host interaction. This review also included a critical analysis of the Latin American context, considering its diverse and unique social, cultural, and biodiversity characteristics. The scarcity of scientific information is Citation: Alvarez-Munoz, S.; striking, thus, a call to local institutions and governments to invest more resources and efforts to the Upegui-Porras, N.; Gomez, A.P.; study of these factors in the region is key. -

Survival of Human Norovirus Surrogates in Juices and Their Inactivation Using Novel Methods
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 5-2011 Survival of Human Norovirus Surrogates In Juices and their Inactivation Using Novel Methods Katie Marie Horm [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Recommended Citation Horm, Katie Marie, "Survival of Human Norovirus Surrogates In Juices and their Inactivation Using Novel Methods. " Master's Thesis, University of Tennessee, 2011. https://trace.tennessee.edu/utk_gradthes/882 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Katie Marie Horm entitled "Survival of Human Norovirus Surrogates In Juices and their Inactivation Using Novel Methods." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Food Science and Technology. Doris H. D'Souza, Major Professor We have read this thesis and recommend its acceptance: Federico M. Harte, Gina M. Pighetti Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Survival of Human Norovirus Surrogates In Juices and their Inactivation Using Novel Methods A Thesis Presented for the Master of Science Degree The University of Tennessee, Knoxville Katie Marie Horm May 2011 Acknowledgments I would like to think my major professor/advisor Dr. -

BK Virus: Current Understanding of Pathogenicity and Clinical Disease in Transplantation
This is a repository copy of BK virus: Current understanding of pathogenicity and clinical disease in transplantation. White Rose Research Online URL for this paper: http://eprints.whiterose.ac.uk/146942/ Version: Accepted Version Article: Chong, S, Antoni, M orcid.org/0000-0002-3641-7559, Macdonald, A orcid.org/0000-0002-5978-4693 et al. (3 more authors) (2019) BK virus: Current understanding of pathogenicity and clinical disease in transplantation. Reviews in Medical Virology, 29 (4). ARTN: e2044. ISSN 1052-9276 https://doi.org/10.1002/rmv.2044 © 2019 John Wiley & Sons, Ltd. This is an author produced version of a paper published in Reviews in Medical Virology. Uploaded in accordance with the publisher's self-archiving policy. Reuse Items deposited in White Rose Research Online are protected by copyright, with all rights reserved unless indicated otherwise. They may be downloaded and/or printed for private study, or other acts as permitted by national copyright laws. The publisher or other rights holders may allow further reproduction and re-use of the full text version. This is indicated by the licence information on the White Rose Research Online record for the item. Takedown If you consider content in White Rose Research Online to be in breach of UK law, please notify us by emailing [email protected] including the URL of the record and the reason for the withdrawal request. [email protected] https://eprints.whiterose.ac.uk/ BK virus: Current understanding of pathogenicity and clinical disease in transplantation Running head: BK Virus: current understanding in Transplantation Stephanie Chong1, Michelle Antoni2, Andrew Macdonald2, Matthew Reeves3, Mark Harber1 and Ciara N. -

Genetic Content and Evolution of Adenoviruses Andrew J
Journal of General Virology (2003), 84, 2895–2908 DOI 10.1099/vir.0.19497-0 Review Genetic content and evolution of adenoviruses Andrew J. Davison,1 Ma´ria Benko´´ 2 and Bala´zs Harrach2 Correspondence 1MRC Virology Unit, Institute of Virology, Church Street, Glasgow G11 5JR, UK Andrew Davison 2Veterinary Medical Research Institute, Hungarian Academy of Sciences, H-1581 Budapest, [email protected] Hungary This review provides an update of the genetic content, phylogeny and evolution of the family Adenoviridae. An appraisal of the condition of adenovirus genomics highlights the need to ensure that public sequence information is interpreted accurately. To this end, all complete genome sequences available have been reannotated. Adenoviruses fall into four recognized genera, plus possibly a fifth, which have apparently evolved with their vertebrate hosts, but have also engaged in a number of interspecies transmission events. Genes inherited by all modern adenoviruses from their common ancestor are located centrally in the genome and are involved in replication and packaging of viral DNA and formation and structure of the virion. Additional niche-specific genes have accumulated in each lineage, mostly near the genome termini. Capture and duplication of genes in the setting of a ‘leader–exon structure’, which results from widespread use of splicing, appear to have been central to adenovirus evolution. The antiquity of the pre-vertebrate lineages that ultimately gave rise to the Adenoviridae is illustrated by morphological similarities between adenoviruses and bacteriophages, and by use of a protein-primed DNA replication strategy by adenoviruses, certain bacteria and bacteriophages, and linear plasmids of fungi and plants. -

Merkel Cell Polyomavirus DNA in Immunocompetent and Immunocompromised Patients with Respiratory Disease
Journal of Medical Virology 83:2220–2224 (2011) Merkel Cell Polyomavirus DNA in Immunocompetent and Immunocompromised Patients With Respiratory Disease Bahman Abedi Kiasari,1,3* Pamela J. Vallely,1 and Paul E. Klapper1,2 1Department of Virology, Genomic Epidemiology Research Group, School of Translational Medicine, University of Manchester, Manchester, United Kingdom 2Clinical Virology, Manchester Medical Microbiology Partnership, Manchester Royal Infirmary, Oxford Road, Manchester, United Kingdom 3Human Viral Vaccine Department, Razi Vaccine & Serum Research Institute, Hesarak, Karaj, Iran Merkel cell polyomavirus (MCPyV) was identi- INTRODUCTION fied originally in association with a rare but aggressive skin cancer, Merkel cell carcinoma. In the past few years, a number of new human poly- The virus has since been found in the respirato- omaviruses, KI, WU, human polyomavirus 6 (HPyV6), ry tract of some patients with respiratory human polyomavirus 7 (HPyV7), trichodysplasia spi- disease. However, the role of MCPyV in the nulosa virus (TSV), human polyomavirus 9 (HPyV9), causation of respiratory disease has not been and Merkel cell polyomavirus (MCPyV) have been established. To determine the prevalence of discovered [Allander et al., 2007; Gaynor et al., 2007; MCPyV in 305 respiratory samples from Feng et al., 2008; Schowalter et al., 2010; van der immunocompetent and immunocompromised Meijden et al., 2010; Scuda et al., 2011]. MCPyV was patients and evaluate their contribution to re- discovered by digital transcriptome subtraction from a spiratory diseases, specimens were screened human skin cancer, Merkel cell carcinoma [Feng for MCPyV using single, multiplex, or real-time et al., 2008]. The finding of MCPyV in human Merkel PCR; co-infection with other viruses was exam- cell carcinoma suggests a role for this virus in the ined. -

Computational Exploration of Virus Diversity on Transcriptomic Datasets
Computational Exploration of Virus Diversity on Transcriptomic Datasets Digitaler Anhang der Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn vorgelegt von Simon Käfer aus Andernach Bonn 2019 Table of Contents 1 Table of Contents 1 Preliminary Work - Phylogenetic Tree Reconstruction 3 1.1 Non-segmented RNA Viruses ........................... 3 1.2 Segmented RNA Viruses ............................. 4 1.3 Flavivirus-like Superfamily ............................ 5 1.4 Picornavirus-like Viruses ............................. 6 1.5 Togavirus-like Superfamily ............................ 7 1.6 Nidovirales-like Viruses .............................. 8 2 TRAVIS - True Positive Details 9 2.1 INSnfrTABRAAPEI-14 .............................. 9 2.2 INSnfrTADRAAPEI-16 .............................. 10 2.3 INSnfrTAIRAAPEI-21 ............................... 11 2.4 INSnfrTAORAAPEI-35 .............................. 13 2.5 INSnfrTATRAAPEI-43 .............................. 14 2.6 INSnfrTBERAAPEI-19 .............................. 15 2.7 INSytvTABRAAPEI-11 .............................. 16 2.8 INSytvTALRAAPEI-35 .............................. 17 2.9 INSytvTBORAAPEI-47 .............................. 18 2.10 INSswpTBBRAAPEI-21 .............................. 19 2.11 INSeqtTAHRAAPEI-88 .............................. 20 2.12 INShkeTCLRAAPEI-44 .............................. 22 2.13 INSeqtTBNRAAPEI-11 .............................. 23 2.14 INSeqtTCJRAAPEI-20 -
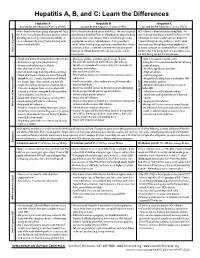
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

On the Coronaviruses and Their Associations with the Aquatic Environment and Wastewater
water Review On the Coronaviruses and Their Associations with the Aquatic Environment and Wastewater Adrian Wartecki 1 and Piotr Rzymski 2,* 1 Faculty of Medicine, Poznan University of Medical Sciences, 60-812 Pozna´n,Poland; [email protected] 2 Department of Environmental Medicine, Poznan University of Medical Sciences, 60-806 Pozna´n,Poland * Correspondence: [email protected] Received: 24 April 2020; Accepted: 2 June 2020; Published: 4 June 2020 Abstract: The outbreak of Coronavirus Disease 2019 (COVID-19), a severe respiratory disease caused by betacoronavirus SARS-CoV-2, in 2019 that further developed into a pandemic has received an unprecedented response from the scientific community and sparked a general research interest into the biology and ecology of Coronaviridae, a family of positive-sense single-stranded RNA viruses. Aquatic environments, lakes, rivers and ponds, are important habitats for bats and birds, which are hosts for various coronavirus species and strains and which shed viral particles in their feces. It is therefore of high interest to fully explore the role that aquatic environments may play in coronavirus spread, including cross-species transmissions. Besides the respiratory tract, coronaviruses pathogenic to humans can also infect the digestive system and be subsequently defecated. Considering this, it is pivotal to understand whether wastewater can play a role in their dissemination, particularly in areas with poor sanitation. This review provides an overview of the taxonomy, molecular biology, natural reservoirs and pathogenicity of coronaviruses; outlines their potential to survive in aquatic environments and wastewater; and demonstrates their association with aquatic biota, mainly waterfowl. It also calls for further, interdisciplinary research in the field of aquatic virology to explore the potential hotspots of coronaviruses in the aquatic environment and the routes through which they may enter it. -

Emerging Viral Diseases of Fish and Shrimp Peter J
Emerging viral diseases of fish and shrimp Peter J. Walker, James R. Winton To cite this version: Peter J. Walker, James R. Winton. Emerging viral diseases of fish and shrimp. Veterinary Research, BioMed Central, 2010, 41 (6), 10.1051/vetres/2010022. hal-00903183 HAL Id: hal-00903183 https://hal.archives-ouvertes.fr/hal-00903183 Submitted on 1 Jan 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Vet. Res. (2010) 41:51 www.vetres.org DOI: 10.1051/vetres/2010022 Ó INRA, EDP Sciences, 2010 Review article Emerging viral diseases of fish and shrimp 1 2 Peter J. WALKER *, James R. WINTON 1 CSIRO Livestock Industries, Australian Animal Health Laboratory (AAHL), 5 Portarlington Road, Geelong, Victoria, Australia 2 USGS Western Fisheries Research Center, 6505 NE 65th Street, Seattle, Washington, USA (Received 7 December 2009; accepted 19 April 2010) Abstract – The rise of aquaculture has been one of the most profound changes in global food production of the past 100 years. Driven by population growth, rising demand for seafood and a levelling of production from capture fisheries, the practice of farming aquatic animals has expanded rapidly to become a major global industry.