UC Santa Barbara Dissertation Template
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rotational Spectroscopy
DAV University Jalandhar Department of Physics Study Material: PHY 512 Atomic & Molecular Spectroscopy M.Sc (Hons.) Physics 2nd sem Source : internet -http://www.phys.ubbcluj.ro/~dana.maniu/OS/BS_3.pdf BOOK: Fundamentals of Molecular Spectroscopy CN Banwell Mc Graw Hill Rotational spectroscopy - Involve transitions between rotational states of the molecules (gaseous state!) - Energy difference between rotational levels of molecules has the same order of magnitude with microwave energy - Rotational spectroscopy is called pure rotational spectroscopy, to distinguish it from roto-vibrational spectroscopy (the molecule changes its state of vibration and rotation simultaneously) and vibronic spectroscopy (the molecule changes its electronic state and vibrational state simultaneously) Molecules do not rotate around an arbitrary axis! Generally, the rotation is around the mass center of the molecule. The rotational axis must allow the conservation of M R α pα const kinetic angular momentum. α Rotational spectroscopy Rotation of diatomic molecule - Classical description Diatomic molecule = a system formed by 2 different masses linked together with a rigid connector (rigid rotor = the bond length is assumed to be fixed!). The system rotation around the mass center is equivalent with the rotation of a particle with the mass μ (reduced mass) around the center of mass. 2 2 2 2 m1m2 2 The moment of inertia: I miri m1r1 m2r2 R R i m1 m2 Moment of inertia (I) is the rotational equivalent of mass (m). Angular velocity () is the equivalent -

(12) United States Patent (10) Patent No.: US 9.468,645 B2 Owoc (45) Date of Patent: Oct
USOO9468645B2 (12) United States Patent (10) Patent No.: US 9.468,645 B2 OWOc (45) Date of Patent: Oct. 18, 2016 (54) HIGHLY SOLUBLE PURINE BOACTIVE (2013.01); A61K 9/0019 (2013.01); A61 K COMPOUNDS AND COMPOSITIONS 9/0095 (2013.01); A61K 9/4858 (2013.01); THEREOF A61 K3I/00 (2013.01); A61K 45/06 (2013.01); A23 V 2002/00 (2013.01) (71) Applicant: JHO Intellectual Property Holdings, (58) Field of Classification Search LLC, Davie, FL (US) CPC ..................... A23V 2002/00; A23V 2200/31; (72) Inventor: Jack Owoc, Weston, FL (US) A23V 2250/062: A23V 2250/21: A23V 2250/5108: A23V 2250/5118: A23V (*) Notice: Subject to any disclaimer, the term of this 2250/54246; A23V 2250/54252: A23V patent is extended or adjusted under 35 2250/704; A23V 2250/712: A61K 31/522; U.S.C. 154(b) by 3 days. A61K 2300/00; A61K 45/06; A61 K9/00 See application file for complete search history. (21) Appl. No.: 14/628,626 (56) References Cited (22) Filed: Feb. 23, 2015 U.S. PATENT DOCUMENTS (65) Prior Publication Data 837,282 A * 12/1906 Owoc ....................... EO1B 9/60 US 2015/O238494 A1 Aug. 27, 2015 238,292 5,973,005 A * 10/1999 D'Amelio, Sr. ... CO7C 277/08 514,565 Related U.S. Application Data 2006/0236421 A1* 10, 2006 Pennell ................ C12N 9/1007 800,278 (60) Provisional application No. 61/944,528, filed on Feb. 2009,0257997 A1* 10, 2009 Owoc .................. A61K9/0014 25, 2014. 424/94.1 (51) Int. C. * cited by examiner A6 IK3I/522 (2006.01) A6 IK 45/06 (2006.01) Primary Examiner – Debbie K Ware A2.3L 2/52 (2006.01) (74) Attorney, Agent, or Firm — David W. -

Identification and Characterization of N 9-Methyltransferase Involved In
ARTICLE https://doi.org/10.1038/s41467-020-15324-7 OPEN Identification and characterization of N9- methyltransferase involved in converting caffeine into non-stimulatory theacrine in tea Yue-Hong Zhang1,2,3,6, Yi-Fang Li1,2,3,6, Yongjin Wang1,3,6, Li Tan1,3, Zhi-Qin Cao1,2,3, Chao Xie1,2,3, Guo Xie4, Hai-Biao Gong1,2,3, Wan-Yang Sun1,2,3, Shu-Hua Ouyang1,2,3, Wen-Jun Duan1,2,3, Xiaoyun Lu1,3, Ke Ding 1,3, ✉ ✉ ✉ ✉ Hiroshi Kurihara1,2,3, Dan Hu 1,2,3 , Zhi-Min Zhang 1,3 , Ikuro Abe 5 & Rong-Rong He 1,2,3 1234567890():,; Caffeine is a major component of xanthine alkaloids and commonly consumed in many popular beverages. Due to its occasional side effects, reduction of caffeine in a natural way is of great importance and economic significance. Recent studies reveal that caffeine can be converted into non-stimulatory theacrine in the rare tea plant Camellia assamica var. kucha (Kucha), which involves oxidation at the C8 and methylation at the N9 positions of caffeine. However, the underlying molecular mechanism remains unclear. Here, we identify the theacrine synthase CkTcS from Kucha, which possesses novel N9-methyltransferase activity using 1,3,7-trimethyluric acid but not caffeine as a substrate, confirming that C8 oxidation takes place prior to N9-methylation. The crystal structure of the CkTcS complex reveals the key residues that are required for the N9-methylation, providing insights into how caffeine N-methyltransferases in tea plants have evolved to catalyze regioselective N-methylation through fine tuning of their active sites. -

Hydrologic Soil Groups
AppendixExhibitAppendix A: Hydrologic AB Soil Synthetic Groups Hydrologic for theRainfall United SoilStates Distributions Groups and Rainfall Data Sources Soils are classified into hydrologic soil groups (HSG’s) Disturbed soil profiles to indicate the minimum rate of infiltration obtained for bareThe highest soil after peak prolonged discharges wetting. from Thesmall HSG watersheds’s, which arein the UnitedAs a result States of areurbanization, usually caused the soil by profileintense, may brief be rain- con- A,falls B, that C, and may D, occur are one as distinctelement eventsused in or determining as part of a longersiderably storm. These altered intense and the rainstorms listed group do not classification usually ex- may runofftended curve over anumbers large area (see and chapter intensities 2). For vary the greatly. conve- One commonno longer practice apply. inIn rainfall-runoffthese circumstances, analysis use is tothe develop follow- niencea synthetic of TR-55 rainfall users, distribution exhibit A-1 to uselists in the lieu HSG of actualclassifi- storming events. to determine This distribution HSG according includes to themaximum texture rainfall of the cationintensities of United for the States selected soils. design frequency arranged in a sequencenew surface that soil, is critical provided for thatproducing significant peak compaction runoff. has not occurred (Brakensiek and Rawls 1983). TheSynthetic infiltration raterainfall is the rate distributions at which water enters the soil at the soil surface. It is controlled by surface condi- HSG Soil textures tions.The length HSG ofalso the indicates most intense the transmission rainfall period rate contributing—the rate to the peak runoff rate is related to the time of concen- A Sand, loamy sand, or sandy loam attration which (T thec) for water the watershed.moves within In thea hydrograph soil. -

Pharmacokinetics of Caffeine: a Systematic Analysis of Reported Data
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.12.452094; this version posted August 3, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Pharmacokinetics of caffeine: A systematic analysis of reported data for application in metabolic phenotyping and liver function testing Jan Grzegorzewski 1, Florian Bartsch 1, Adrian Koller¨ 1, and Matthias Konig¨ 1;∗ 1Institute for Theoretical Biology, Humboldt-University Berlin, Invalidenstraße 110, 10115 Berlin, Germany Correspondence*: Matthias Konig¨ [email protected] ABSTRACT Caffeine is by far the most ubiquitous psychostimulant worldwide found in tea, coffee, cocoa, energy drinks, and many other beverages and food. Caffeine is almost exclusively metabolized in the liver by the cytochrome P-450 enzyme system to the main product paraxanthine and the additional products theobromine and theophylline. Besides its stimulating properties, two important applications of caffeine are metabolic phenotyping of cytochrome P450 1A2 (CYP1A2) and liver function testing. An open challenge in this context is to identify underlying causes of the large inter-individual variability in caffeine pharmacokinetics. Data is urgently needed to understand and quantify confounding factors such as lifestyle (e.g. smoking), the effects of drug-caffeine interactions (e.g. medication metabolized via CYP1A2), and the effect of disease. Here we report the first integrative and systematic analysis of data on caffeine pharmacokinetics from 148 publications and provide a comprehensive high-quality data set on the pharmacokinetics of caffeine, caffeine metabolites, and their metabolic ratios in human adults. The data set is enriched by meta-data on the characteristics of studied patient cohorts and subjects (e.g. -

Caffeine and Chlorogenic Acid Contents Among Wild Coffea
Food Chemistry Food Chemistry 93 (2005) 135–139 www.elsevier.com/locate/foodchem Qualitative relationship between caffeine and chlorogenic acid contents among wild Coffea species C. Campa, S. Doulbeau, S. Dussert, S. Hamon, M. Noirot * Centre IRD of Montpellier, B.P. 64501, 34394 Montpellier Cedex 5, France Received 28 June 2004; received in revised form 13 October 2004; accepted 13 October 2004 Abstract Chlorogenic acids, sensu largo (CGA), are secondary metabolites of great economic importance in coffee: their accumulation in green beans contributes to coffee drink bitterness. Previous evaluations have already focussed on wild species of coffee trees, but this assessment included six new taxa from Cameroon and Congo and involved a simplified method that generated more accurate results. Five main results were obtained: (1) Cameroon and Congo were found to be a centre of diversity, encompassing the entire range of CGA content from 0.8% to 11.9% dry matter basis (dmb); (2) three groups of coffee tree species – CGA1, CGA2 and CGA3 – were established on the basis of discontinuities; (3) means were 1.4%, 5.6% and 9.9% dmb, respectively; (4) there was a qualitative relation- ship between caffeine and ACG content distribution; (5) only a small part of the CGA is trapped by caffeine as caffeine chlorogenate. Ó 2004 Elsevier Ltd. All rights reserved. Keywords: Coffeae; Caffeine; Chlorogenic acids 1. Introduction lic acid [feruloylquinic acids (FQA)] and represent 98% of all CGAs (Clifford, 1985; Morishita, Iwahashi, & Two coffee species, C. canephora and C. arabica, are Kido, 1989). There are also some minor compounds, cultivated worldwide. However, these species only repre- such as esters of feruloyl-caffeoylquinic acids (FCQA), sent a small proportion of the worldÕs coffee genetic re- caffeoyl-feruloylquinic acids (CFQA) or p-coumaric acid sources as, in the wild, the Coffea subgenus (Coffea (p-CoQA). -

Theacrine Attenuates Myocardial Fibrosis After Myocardial Infarction Via the SIRT3/Β-Catenin/ Pparγ Pathway in Estrogen-Deficient Mice
European Review for Medical and Pharmacological Sciences 2019; 23: 5477-5486 Theacrine attenuates myocardial fibrosis after myocardial infarction via the SIRT3/β-catenin/ PPARγ pathway in estrogen-deficient mice L.-L. SONG1, Y. ZHANG2, X.-R. ZHANG3, Y.-N. SONG4, H.-Z. DAI5 1Department of Critical Care Medicine, Weifang Hanting District People’s Hospital, Weifang, China 2Department of Respiratory Medicine, Weifang Hanting District People’s Hospital, Weifang, China 3Department of Pharmacology, Weifang Medical University, Weifang, China 4Department of Breast and Thyroid Surgery, The Traditional Chinese Medicine Hospital of Weifang, Weifang, China 5Department of Emergency, The Affiliated Hospital of Weifang Medical University, Weifang, China Abstract. – OBJECTIVE: To investigate the myocardial apoptosis were clearly aggravated, role of theacrine in the protection of ventricular and the SIRT3 expression was decreased at 28 remodeling and chronic heart failure after myo- days after myocardial infarction. The theacrine cardial infarction in the estrogen-deficient mice. could improve the cardiac function after the MATERIALS AND METHODS: Female myocardial infarction in estrogen-deficient mice C57BL/6 mice aged 8 weeks old were selected and relieve both myocardial fibrosis and myo- and then subjected to bilateral oophorectomy. cardial apoptosis during chronic remodeling af- At 7 days after surgery, the models of the myo- ter myocardial infarction in estrogen-deficient cardial infarction were established by ligating mice. After the intervention with theacrine, the the anterior descending coronary artery. On the estrogen-deficient mice with myocardial infarc- first day after myocardial infarction, Theacrine tion had up-regulated SIRT3 and PPARγ levels (20 mg/kg) was administered via gavage for con- and a reduced β-catenin level in the heart. -

Herbal Principles in Cosmetics Properties and Mechanisms of Action Traditional Herbal Medicines for Modern Times
Traditional Herbal Medicines for Modern Times Herbal Principles in Cosmetics Properties and Mechanisms of Action Traditional Herbal Medicines for Modern Times Each volume in this series provides academia, health sciences, and the herbal medicines industry with in-depth coverage of the herbal remedies for infectious diseases, certain medical conditions, or the plant medicines of a particular country. Series Editor: Dr. Roland Hardman Volume 1 Shengmai San, edited by Kam-Ming Ko Volume 2 Rasayana: Ayurvedic Herbs for Rejuvenation and Longevity, by H.S. Puri Volume 3 Sho-Saiko-To: (Xiao-Chai-Hu-Tang) Scientific Evaluation and Clinical Applications, by Yukio Ogihara and Masaki Aburada Volume 4 Traditional Medicinal Plants and Malaria, edited by Merlin Willcox, Gerard Bodeker, and Philippe Rasoanaivo Volume 5 Juzen-taiho-to (Shi-Quan-Da-Bu-Tang): Scientific Evaluation and Clinical Applications, edited by Haruki Yamada and Ikuo Saiki Volume 6 Traditional Medicines for Modern Times: Antidiabetic Plants, edited by Amala Soumyanath Volume 7 Bupleurum Species: Scientific Evaluation and Clinical Applications, edited by Sheng-Li Pan Traditional Herbal Medicines for Modern Times Herbal Principles in Cosmetics Properties and Mechanisms of Action Bruno Burlando, Luisella Verotta, Laura Cornara, and Elisa Bottini-Massa Cover art design by Carlo Del Vecchio. CRC Press Taylor & Francis Group 6000 Broken Sound Parkway NW, Suite 300 Boca Raton, FL 33487-2742 © 2010 by Taylor and Francis Group, LLC CRC Press is an imprint of Taylor & Francis Group, an Informa business No claim to original U.S. Government works Printed in the United States of America on acid-free paper 10 9 8 7 6 5 4 3 2 1 International Standard Book Number-13: 978-1-4398-1214-3 (Ebook-PDF) This book contains information obtained from authentic and highly regarded sources. -

The Two Tortoitumia Ultimate Tatli Himin
THETWO TORTOITUMIAUS 20180104248A1ULTIMATE TATLI HIMIN ( 19) United States ( 12) Patent Application Publication ( 10 ) Pub . No. : US 2018 /0104248 A1 Lopez et al. (43 ) Pub . Date : Apr . 19 , 2018 ( 54 ) THEACRINE -BASED SUPPLEMENT AND Publication Classification METHOD OF USE THEREOF IN A (51 ) Int. Cl. SYNERGISTIC COMBINATION WITH A61K 31/ 522 ( 2006 . 01 ) CAFFEINE C07D 487/ 04 (2006 . 01) (52 ) U . S . CI. @( 71 ) Applicant : Ortho - Nutra, LLC , Morganville , NJ CPC .. A61K 31/ 522 ( 2013 .01 ) ; C07D 487 /04 (US ) ( 2013. 01 ) @(72 ) Inventors : Hector L Lopez , Cream Ridge , NJ (US ) ; Shawn Wells , Lewisville , TX (US ) ; Tim N . Ziegenfuss , Chardon , OH (57 ) ABSTRACT (US ) A human dietary supplement comprises theacrine and @( 73 ) Assignee : Ortho -Nutra , LLC , Morganville , NJ optionally other compounds that modulate the effects of theacrine . Uses for the theacrine - containing supplement (US ) include improvement of at least one ofmood , energy, focus, concentration or sexual desire or a reduction of at least one (21 ) Appl. No. : 15 /600 , 371 of anxiety or fatigue. A synergistic composition comprises ( 22 ) Filed : May 19 , 2017 co -administration of theacrine and caffeine , wherein the co - administered caffeine reduces theacrine oral clearance Related U . S . Application Data (CL / F ) and oral volume of distribution (Vd / F ) . In addition , the co - administered caffeine increases area under the plasma (63 ) Continuation - in -part of application No. 14 / 539, 726 , concentration time curve ( AUC ) of theacrine , and increases filed on Nov. 12 , 2014 . theacrine maximum plasma concentration ( Cmax ) in com (60 ) Provisional application No .61 / 903 ,362 , filed on Nov . parison with the corresponding pharmacokinetic parameters 12 , 2013 . -

UC Santa Barbara Dissertation Template
UNIVERSITY OF CALIFORNIA Santa Barbara Laser Spectroscopy and Photodynamics of Alternative Nucleobases and Organic Dyes A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Chemistry by Jacob Alan Berenbeim Committee in charge: Professor Mattanjah de Vries, Chair Professor Steve Buratto Professor Michael Gordon Professor Martin Moskovits December 2017 The dissertation of Jacob Alan Berenbeim is approved. ____________________________________________ Steve Buratto ____________________________________________ Michael Gordon ____________________________________________ Martin Moskovits ____________________________________________ Mattanjah de Vries, Committee Chair October 2017 Laser Spectroscopy and Photodynamics of Alternative Nucleobases and Organic Dyes Copyright © 2017 by Jacob Alan Berenbeim iii ACKNOWLEDGEMENTS To my wife Amy thank you for your endless support and for inspiring me to match your own relentless drive towards reaching our goals. To my parents and my brothers Eli and Gabe thank you for your love and visits to Santa Barbara, CA. To my advisor Mattanjah and my lab mates thank you for the incredible opportunity to share ideas and play puppets with the fabric of space. And to my cat Lola, you’re a good cat. iv VITA OF JACOB ALAN BERENBEIM October 2017 EDUCATION University of California, Santa Barbara CA Fall 2017 PhD, Physical Chemistry Advisor: Prof. Mattanjah S. de Vries University of Puget Sound, Tacoma WA 2009 BS, Chemistry Advisor: Prof. Daniel Burgard LABORATORY TECHNIQUES Photophysics by UV/VIS and IR pulsed laser spectroscopy, optical alignment, oa-TOF mass spectrometry (multiphoton ionization, MALDI, ESI+), molecular beam high vacuum apparatus, high voltage electronics, molecular computational modeling with Gaussian, data acquisition with LabView, and data manipulation with Mathematica and Origin RESEARCH EXPERIENCE Graduate Student Researcher 2012-2017 • Time dependent (transient) photo relaxation of organic molecules, including PAHs and aromatic biological molecules. -
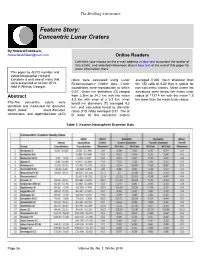
Concentric Lunar Craters
The Strolling Astronomer Feature Story: Concentric Lunar Craters By Howard Eskildsen, [email protected] Online Readers Left-click your mouse on the e-mail address in blue text to contact the author of this article, and selected references also in blue text at the end of this paper for more information there. This paper by ALPO member and astrophotographer Howard Eskidsen is only one of many that ratios were calculated using Lunar averaged 0.065, much shallower than were presented at ALCon 2013, Reconnaissance Orbiter data. Crater the T/D ratio of 0.20 that is typical for held in Atlanta, Georgia. coordinates were reproducible to within non-concentric craters. Mean crater rim 0.02°. Outer rim diameters (D) ranged elevations were below the mean lunar Abstract from 2.3km to 24.2 km with the mean radius of 1737.4 km with the mean 1.5 8.2 km with error of ± 0.2 km. Inner km lower than the mean lunar radius. Fifty-five concentric caters were toroid rim diameters (T) averaged 4.3 identified and measured for diameter, km, and calculated toroid to diameter depth, toroid crest diameter, ratios (T/D ratio) averaged 0.51. The d/ coordinates, and depth/diameter (d/D) D ratios for the concentric craters Table 1: Eastern Hemisphere Diameter Data Page 36 Volume 56, No. 1, Winter 2014 The Strolling Astronomer Table 1A: Eastern Hemisphere Elevation/Depth Data Concentric Craters pure highland areas. None are found in While the most notable concentrics have A small percentage of craters that would the central maria (Wood 1978). -

Tuesday Morning, October 16, 2007
Tuesday Morning, October 16, 2007 Electronic Materials and Processing 9:20am EM-TuM5 Real-time Conductivity Analysis through Single- Molecule Electrical Junctions, J.-S. Na, J. Ayres, K.L. Chandra, C.B. Room: 612 - Session EM-TuM Gorman, G.N. Parsons, North Carolina State University We have recently developed a molecular electronic characterization test-bed Molecular Electronics that utilizes a symmetric pair of gold nanoparticles, 40 nm in diameter, joined together by a single or small group of conjugated oligomeric phenylene ethynylene (OPE) molecules. These Moderator: I. Hill, Dalhousie University nanoparticle/molecule/nanoparticle structures are subsequently assembled between nanoscale test electrodes to enable current through the molecule to 8:00am EM-TuM1 Dynamics of Molecular Switch Molecules Imaged be characterized. At low voltage (< ±1.5 V) the observed current is by Alternating Current Scanning Tunneling Microscopy, A.M. Moore, consistent with common non-resonant tunneling (i.e., I vs V is independent P.S. Weiss, The Pennsylvania State University of temperature between 80 and 300 K). This molecular analysis approach is We have studied oligo(phenylene-ethylene) (OPE) molecules as candidates unique because it enables the stability of the molecular conductance to be for molecular electronic switches. We previously determined the switching observed and characterized over extended periods (several weeks so far) mechanism to rely on hybridization changes between the substrate and after fabrication. Conductance through single molecule junctions was molecule. Here, we have determined which molecules will be more or less monitored in real-time during several process sequences, including active in our samples using a custom-built alternating current scanning dielectrophoretic directed self assembly and post-assembly modification.