Bsc Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

George A. Olah 151
MY SEARCH FOR CARBOCATIONS AND THEIR ROLE IN CHEMISTRY Nobel Lecture, December 8, 1994 by G EORGE A. O L A H Loker Hydrocarbon Research Institute and Department of Chemistry, University of Southern California, Los Angeles, CA 90089-1661, USA “Every generation of scientific men (i.e. scientists) starts where the previous generation left off; and the most advanced discov- eries of one age constitute elementary axioms of the next. - - - Aldous Huxley INTRODUCTION Hydrocarbons are compounds of the elements carbon and hydrogen. They make up natural gas and oil and thus are essential for our modern life. Burning of hydrocarbons is used to generate energy in our power plants and heat our homes. Derived gasoline and diesel oil propel our cars, trucks, air- planes. Hydrocarbons are also the feed-stock for practically every man-made material from plastics to pharmaceuticals. What nature is giving us needs, however, to be processed and modified. We will eventually also need to make hydrocarbons ourselves, as our natural resources are depleted. Many of the used processes are acid catalyzed involving chemical reactions proceeding through positive ion intermediates. Consequently, the knowledge of these intermediates and their chemistry is of substantial significance both as fun- damental, as well as practical science. Carbocations are the positive ions of carbon compounds. It was in 1901 that Norris la and Kehrman lb independently discovered that colorless triphe- nylmethyl alcohol gave deep yellow solutions in concentrated sulfuric acid. Triphenylmethyl chloride similarly formed orange complexes with alumi- num and tin chlorides. von Baeyer (Nobel Prize, 1905) should be credited for having recognized in 1902 the salt like character of the compounds for- med (equation 1). -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic Substitution
Dr. Tripti Gangwar AROMATIC NUCLEOPHILIC SUBSTITUTION-PART -2 Electrophilic substitution ◦ The aromatic ring acts as a nucleophile, and attacks an added electrophile E+ ◦ An electron-deficient carbocation intermediate is formed (the rate- determining step) which is then deprotonated to restore aromaticity ◦ electron-donating groups on the aromatic ring (such as -OH, -OCH3, and alkyl) make the reaction faster, since they help to stabilize the electron-poor carbocation intermediate ◦ Lewis acids can make electrophiles even more electron-poor (reactive), increasing the reaction rate. For example FeBr3 / Br2 allows bromination to occur at a useful rate on benzene, whereas Br2 by itself is slow). In fact, a substitution reaction does occur! (But, as you may suspect, this isn’t an electrophilic aromatic substitution reaction.) In this substitution reaction the C-Cl bond breaks, and a C-O bond forms on the same carbon. The species that attacks the ring is a nucleophile, not an electrophile The aromatic ring is electron-poor (electrophilic), not electron rich (nucleophilic) The “leaving group” is chlorine, not H+ The position where the nucleophile attacks is determined by where the leaving group is, not by electronic and steric factors (i.e. no mix of ortho– and para- products as with electrophilic aromatic substitution). In short, the roles of the aromatic ring and attacking species are reversed! The attacking species (CH3O–) is the nucleophile, and the ring is the electrophile. Since the nucleophile is the attacking species, this type of reaction has come to be known as nucleophilic aromatic substitution. n nucleophilic aromatic substitution (NAS), all the trends you learned in electrophilic aromatic substitution operate, but in reverse. -

19.1 Ketones and Aldehydes
19.1 Ketones and Aldehydes • Both functional groups possess the carbonyl group • Important in both biology and industry Simplest aldehyde Simplest ketone used as a used mainly as preservative a solvent Copyright © 2017 John Wiley & Sons, Inc. All rights reserved. 19-1 Klein, Organic Chemistry 3e 19.1 Ketones And Aldehydes Copyright © 2017 John Wiley & Sons, Inc. All rights reserved. 19-2 Klein, Organic Chemistry 3e 19.2 Nomenclature • Four discrete steps to naming an aldehyde or ketone • Same procedure as with alkanes, alcohols, etc… 1. Identify and name the parent chain 2. Identify the name of the substituents (side groups) 3. Assign a locant (number) to each substituents 4. Assemble the name alphabetically Copyright © 2017 John Wiley & Sons, Inc. All rights reserved. 19-3 Klein, Organic Chemistry 3e 19.2 Nomenclature 1. Identify and name the parent chain – For aldehydes, replace the “-e” ending with an “-al” – the parent chain must include the carbonyl carbon Copyright © 2017 John Wiley & Sons, Inc. All rights reserved. 19-4 Klein, Organic Chemistry 3e 19.2 Nomenclature 1. Identify and name the parent chain – The aldehydic carbon is assigned number 1: Copyright © 2017 John Wiley & Sons, Inc. All rights reserved. 19-5 Klein, Organic Chemistry 3e 19.2 Nomenclature 1. Identify and name the parent chain – For ketones, replace the “-e” ending with an “-one” – The parent chain must include the C=O group – the C=O carbon is given the lowest #, and can be expressed before the parent name or before the suffix Copyright © 2017 John Wiley & Sons, Inc. All rights reserved. -

Aromatic Nucleophilic Substitution Reaction
Aromatic Nucleophilic Substitution Reaction DR. RAJENDRA R TAYADE ASSISTANT PROFESSOR DEPARTMENT OF CHEMISTRY INSTITUTE OF SCIENCE, NAGPUR Principles There are four principal mechanisms for aromatic nucleophilic substitution which are similar to that of aliphatic nucleophilic substitution. (SN1, SN2, SNi, SET Mechanism) 1. SNAr Mechanism- addition / elimination CF3, CN, CHO, COR, COOH, Br, Cl, I Common Activating Groups for NAS Step [1] Addition of the nucleophile (:Nu–) to form a carbanion Addition of the nucleophile (:Nu–) forms a resonance-stabilized carbanion with a new C – Nu bond— three resonance structures can be drawn. • Step is rate-determining • Aromaticity of the benzene ring is lost Step [2] loss of the leaving group re-forms the aromatic ring. • This step is fast because the aromaticity of the benzene ring is restored. ? Explain why a methoxy group (CH3O) increases the rate of electrophilic aromatic substitution, but decreases the rate of nucleophilic aromatic substitution. 2.ArSN1 Mechanism- elimination /addition • This mechanism operates in the reaction of diazonium salts with nucleophiles. •The driving force resides in the strength of the bonding in the nitrogen molecule that makes it a particularly good leaving group. 3.Benzyne Mechanism- elimination /addition Step [1] Elimination of HX to form benzyne Elimination of H and X from two adjacent carbons forms a reactive benzyne intermediate Step [2] Nucleophilic addition to form the substitution product Addition of the nucleophile (–OH in this case) and protonation form the substitution product Evidence for the Benzyne Mechanism Trapping in Diels/Alder Reaction O O B E N Z Y N E C C O O O D i e l s / A l d e r O N H 3 N N Dienophile Diene A d d u c t Substrate Modification – absence of a hydrogens LG Substituent Substituent No Reaction Base Isotopic Labeling LG Nu H Nu Structure of Benzyne • The σ bond is formed by overlap of two sp2 hybrid orbitals. -

A Marcus-Theory-Based Approach to Ambident Reactivity
Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München A Marcus-Theory-Based Approach to Ambident Reactivity Robert Martin Breugst aus München 2010 Erklärung Diese Dissertation wurde im Sinne von § 13 Abs. 3 bzw. 4 der Promotionsordnung vom 29. Januar 1998 (in der Fassung der vierten Änderungssatzung vom 26. November 2004) von Herrn Professor Dr. Herbert Mayr betreut. Ehrenwörtliche Versicherung Diese Dissertation wurde selbständig, ohne unerlaubte Hilfe erarbeitet. München, 28.10.2010 _________________________ Dissertation eingereicht am: 28.10.2010 1. Gutachter: Prof. Dr. Herbert Mayr 2. Gutachter: Prof. Dr. Hendrik Zipse Mündliche Prüfung am: 21.12.2010 II Acknowledgements Acknowledgements First of all, I would like to express my cordial gratitude to Professor Dr. Herbert Mayr for the opportunity to compose this thesis in his group. I have cherished all the valuable discussions with him, his endless support, and his inspiring confidence very much and I have always appreciated working under these excellent conditions. From the very first day in his group Professor Mayr made me feel welcome, has always been willing to discuss my ideas, and encouraged me to pursue a scientific career. Furthermore, I would like to thank Professor Dr. Hendrik Zipse for numerous discussions and helpful comments on quantum chemical calculations, and of course also for reviewing my thesis. Additionally, I am very indebted to Professor J. Peter Guthrie, Ph.D., for giving me the opportunity to spend almost 4 months in his group at the University of Western Ontario. During this time, I have learned so many new things and I am grateful for the reams of discussions in his office. -
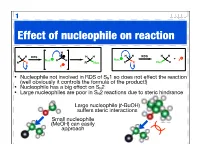
Effect of Nucleophile on Reaction
1 Effect of nucleophile on reaction H H H H H H H RDS H H RDS + Nuc R H Nuc X R X R Nuc R X Nuc R X • Nucleophile not involved in RDS of SN1 so does not effect the reaction (well obviously it controls the formula of the product!) • Nucleophile has a big effect on SN2 • Large nucleophiles are poor in SN2 reactions due to steric hindrance Large nucleophile (t-BuOH) suffers steric interactions Small nucleophile (MeOH) can easily approach 2 Nucleophile strength • The stronger the nucleophile the faster / more efficient the SN2 reaction • Nucleophilic strength (nucleophilicity) relates to how easily a compound can donate an electron pair • The more electronegative an atom the less nucleophilic as the electrons are held closer nucleophilic strength Anion more nucleophilic H O than its neutral analogue (basicity) HO > 2 nucleophilic strength R C (basicity) 3 > R2N > RO > F As we move along a row the electronegativity electronegativity C < N < O < F increases and nucleophilic strength nucleophilicity decreases (basicity) R NH2 > R OH > R F As we go down a group both anions and neutral atoms get more nucleophilic strength H2S > H2O (basicity) nucleophilic - electrons I > Br > Cl > F not held as tightly as size increases 3 Solvent effects SN1 • Intermediate - the carbocation - is stabilised by polar solvents • Leaving group stabilised by protic solvents (encourages dissociation) • Water, alcohols & carboxylic acids good solvents for SN1 Br H O H O H H Br O H O H O cation hydrogen stabilised O bonds SN2 • Prefer less polar, aprotic solvents • Less -

CHEMICAL HERITAGE FOUNDATION GEORGE A. OLAH Transcript
CHEMICAL HERITAGE FOUNDATION GEORGE A. OLAH Transcript of an Interview Conducted by James G. Traynham and Arnold Thackray at Loker Hydrocarbon Research Institute Los Angeles, California on 3 February 2000 (With Subsequent Corrections and Additions) This interview has been designated as Free Access. One may view, quote from, cite, or reproduce the oral history with the permission of CHF. Please note: Users citing this interview for purposes of publication are obliged under the terms of the Chemical Heritage Foundation Oral History Program to credit CHF using the format below: George A. Olah, interview by James G. Traynham and Arnold Thackray at Loker Hydrocarbon Research Institute, Los Angeles, California, 3 February 2000 (Philadelphia: Chemical Heritage Foundation, Oral History Transcript # 0190). Chemical Heritage Foundation Oral History Program 315 Chestnut Street Philadelphia, Pennsylvania 19106 GEORGE A. OLAH 1927 Born in Budapest, Hungary, on 22 May Education 1945 B.S., organic chemistry, Technical University, Budapest 1949 Ph.D., organic chemistry, Technical University, Budapest Professional Experience Technical University, Budapest, Hungary 1949-1954 Assistant Professor to Associate Professor of Organic Chemistry Hungarian Academy of Sciences 1954-1956 Head of Department of Organic Chemistry and Associate Scientific Director of Central Research Institute The Dow Chemical Company 1957-1964 Senior Research Scientist Case Western Reserve University 1965-1967 Professor and Chairman, Department of Chemistry 1967-1969 Chairman of Combined Departments of Chemistry (Case Institute and Western Reserve University) 1967-1977 C. F. Mabery Distinguished Professor of Research in Chemistry University of Southern California 1977-1977 Professor of Chemistry and Scientific Director, Hydrocarbon Research Institute 1980-present Distinguished Professor of Chemistry 1983-present Donald P. -

Reactions of Alkenes Since Bonds Are Stronger Than Bonds, Double Bonds Tend to React to Convert the Double Bond Into Bonds
Reactions of Alkenes Since bonds are stronger than bonds, double bonds tend to react to convert the double bond into bonds This is an addition reaction. (Other types of reaction have been substitution and elimination). Addition reactions are typically exothermic. Electrophilic Addition The bond is localized above and below the C-C bond. The electrons are relatively far away from the nuclei and are therefore loosely bound. An electrophile will attract those electrons, and can pull them away to form a new bond. This leaves one carbon with only 3 bonds and a +ve charge (carbocation). The double bond acts as a nucleophile (attacks the electrophile). Ch08 Reacns of Alkenes (landscape) Page 1 In most cases, the cation produced will react with another nucleophile to produce the final overall electrophilic addition product. Electrophilic addition is probably the most common reaction of alkenes. Consider the electrophilic addition of H-Br to but-2-ene: The alkene abstracts a proton from the HBr, and a carbocation and bromide ion are generated. The bromide ion quickly attacks the cationic center and yields the final product. In the final product, H-Br has been added across the double bond. Ch08 Reacns of Alkenes (landscape) Page 2 Orientation of Addition Consider the addition of H-Br to 2-methylbut-2-ene: There are two possible products arising from the two different ways of adding H-Br across the double bond. But only one is observed. The observed product is the one resulting from the more stable carbocation intermediate. Tertiary carbocations are more stable than secondary. -

Elimination Reaction
Elimination reaction An elimination reaction is a type of organic reaction in which two substituents are removed from a molecule in either one or two-step mechanism.[2] The one-step mechanism is known as the E2 reaction, and the two-step mechanism is known as the E1 reaction. The numbers refer not to the number of steps in the mechanism, but rather to the kinetics of the reaction: E2 is Elimination reaction of cyclohexanol to bimolecular (second-order) while E1 is unimolecular (first-order). cyclohexene with sulfuric acid and In cases where the molecule is able to stabilize an anion but heat[1] possesses a poor leaving group, a third type of reaction, E1CB, exists. Finally, the pyrolysis of xanthate and acetate esters proceed through an "internal" elimination mechanism, the Ei mechanism. Contents Loss of hydron (H+) E2 mechanism E1 mechanism Competition among mechanisms Elimination reactions other than β-elimination See also References External links Loss of hydron (H+) In most organic elimination reactions, at least one hydron (H+) is lost to form the double bond: the unsaturation of the molecule increases. It is also possible that a molecule undergoes reductive elimination, by which the valence of an atom in the molecule decreases by two, though this is more common in inorganic chemistry. An important class of elimination reactions is those involving alkyl halides, with good leaving groups, reacting with a Lewis base to form an alkene. Elimination may be considered the reverse of an addition reaction. When the substrate is asymmetric, regioselectivity is determined by Zaitsev's rule or through Hofmann elimination if the carbon with the most substituted hydrogen is inaccessible. -
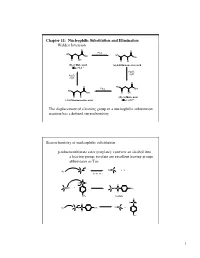
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion
Chapter 11: Nucleophilic Substitution and Elimination Walden Inversion O O PCl5 HO HO OH OH O OH O Cl (S)-(-) Malic acid (+)-2-Chlorosuccinic acid [a]D= -2.3 ° Ag2O, H2O Ag2O, H2O O O HO PCl5 OH HO OH O OH O Cl (R)-(+) Malic acid (-)-2-Chlorosuccinic acid [a]D= +2.3 ° The displacement of a leaving group in a nucleophilic substitution reaction has a defined stereochemistry Stereochemistry of nucleophilic substitution p-toluenesulfonate ester (tosylate): converts an alcohol into a leaving group; tosylate are excellent leaving groups. abbreviates as Tos C X Nu C + X- Nu: X= Cl, Br, I O Cl S O O + C OH C O S CH3 O CH3 tosylate O -O S O O Nu C + Nu: C O S CH3 O CH3 1 O Tos-Cl - H3C O O H + TosO - H O H pyridine H O Tos O CH3 [a]D= +33.0 [a]D= +31.1 [a]D= -7.06 HO- HO- O - Tos-Cl H3C O - H O O TosO + H O H pyridine Tos H O CH3 [a]D= -7.0 [a]D= -31.0 [a]D= -33.2 The nucleophilic substitution reaction “inverts” the Stereochemistry of the carbon (electrophile)- Walden inversion Kinetics of nucleophilic substitution Reaction rate: how fast (or slow) reactants are converted into product (kinetics) Reaction rates are dependent upon the concentration of the reactants. (reactions rely on molecular collisions) H H Consider: HO C _ _ C Br Br HO H H H H At a given temperature: If [OH-] is doubled, then the reaction rate may be doubled If [CH3-Br] is doubled, then the reaction rate may be doubled A linear dependence of rate on the concentration of two reactants is called a second-order reaction (molecularity) 2 H H HO C _ _ C Br Br HO H H H H Reaction rates (kinetic) can be expressed mathematically: reaction rate = disappearance of reactants (or appearance of products) For the disappearance of reactants: - rate = k [CH3Br] [OH ] [CH3Br] = CH3Br concentration [OH-] = OH- concentration k= constant (rate constant) L mol•sec For the reaction above, product formation involves a collision between both reactants, thus the rate of the reaction is dependent upon the concentration of both.