A Multicenter Phase I Study Evaluating Dual PI3K and BRAF Inhibition with PX-866
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Yoga in the Complementary Treatment of Cancer
MOJ Yoga & Physical Therapy Mini Review Open Access The role of Yoga in the complementary treatment of cancer Abstract Volume 2 Issue 3 - 2017 The life of a cancer patient is complicated by a litany of physical, psychological, social 1 2 and spiritual factors leading to anxiety, fatigue, depression and several other unpleasant Neil K Agarwal, Shashi K Agarwal 1 emotional issues. Nausea and vomiting, insomnia and pain also contribute greatly to Hahnemann University Hospital, USA 2 the overall discomfort. These symptoms often result in a significant reduction in the Center for Contemporary and Complementary Cardiology, USA quality of life. A host of non-pharmacological therapeutic interventions have been tried to alleviate this associated physical and emotional issues in cancer patients, with Correspondence: Neil K Agarwal, Hahnemann University limited success. Yoga therapy has increasingly demonstrated evidence based benefits Hospital, USA, Email [email protected] in alleviating many of these cancer-related symptoms and in greatly improving the quality of life of these patients. Received: May 10, 2017 | Published: September 18, 2017 Keywords: yoga, cancer, anxiety, depression, fatigue, nausea and vomiting, cancer pain, quality of life Introduction Results Yoga evolved over thousands of years in India. The ancient sages Search under ‘yoga and cancer’ revealed 339 citations dating developed this practice as an integrative physical, psychological and back to 1975 on PubMed. PMC revealed 2,736 full length articles. spiritual regimen -

BARWON HEALTH RESEARCH REPORT 2018 Foreword
Research Report 20 18 Contents 4 Foreword 36 Endocrinology 108 Opthamology 40 Epidemiology (EPI-Centre 109 Oral Health Services Section 1 for Healthy Ageing) 6 112 Palliative Care Overview 45 Infectious Diseases 114 Pharmacy 8 Academic Strategic Plan 48 Nursing 117 Physiotherapy 10 The Barwon Health Foundation 54 Nutritional Psychiatry (Food 118 Social Work and Future Fund And Mood Centre) 120 Speech Pathology 12 St Mary’s Library and 59 Orthopaedic Surgery Research Centre 122 Urology 63 Paediatrics (The Child Health 13 Grants Received Research Unit (CHRU) Section 5 14 Career Spotlight - Professor 65 Psychiatry (Impact SRC) 124 Mark Kotowicz Clinical Trials 78 Surgery 126 Clinical Trials Advisory 84 Career Spotlight - 16 Section 2 Committee Summary Dr Paul Talman Research Directorate And (CTAC) 2018 Barwon Health Human 128 Cardiology Research Ethics Committee 86 Section 4 130 Endocrinology/Infectious (HREC) Barwon Health Research Diseases And Pediatrics 18 Barwon Health Research Roundups 132 IMPACT Directorate 88 Aged Care 134 Intensive Care 22 Barwon Health Human 89 Anaesthesia Research Ethics 138 Oncology And Haematology Committee (HREC) 91 Barwon Medical Imaging (BMI) 25 Research Week 2017 Summary 93 Cancer Services Section 6 142 And Outcomes 96 Cardiology A Snapshot Of Australian 26 Career Spotlight - 98 Community Health Conferences Dr Greg Weeks 99 Healthlinks And 160 A Snapshot Of International Personalised Care Conferences 28 Section 3 101 Hospital Admission Risk Barwon Health/Deakin Program (HARP) 164 Section 7 University Collaborative Publications Research Groups 102 Nephrology 30 Emerging Infectious 105 Occupational Therapy Diseases (GCEID) 3 BARWON HEALTH RESEARCH REPORT 2018 Foreword Welcome to the 2018 Research Report, Barwon Health’s fourth annual edition. -

050125Understanding Nausea and Vomiting in Advanced Cancer
CLINICAL knowledge Understanding nausea and vomiting in advanced cancer Author Colin Perdue, Bn, Rgn, dipn, is clinical nurse Definitions specialist in palliative medicine, Morriston Hospital, Nausea and vomiting are often regarded as a single Swansea. entity, but they are separate physiological conditions AbstrAct Perdue, C. (2005) Understanding nausea (Eckert, 2001). Nausea is an unpleasant feeling of the and vomiting in advanced cancer. Nursing Times; 101, need to vomit. It is often accompanied by autonomic 4, 32–35. symptoms such as pallor, cold sweat, salivation and tachy- Nausea and vomiting are commonly experienced by cardia (Yarbro et al, 1999). Retching is a strong involuntary people with advanced cancer. Nausea and vomiting can effort to vomit. It occurs in the presence of nausea and RefeRences have an adverse effect on a patient’s physical, psycho- often culminates in vomiting (Twycross and Back, 1998). logical and social well-being. Knowledge of the physiol- Vomiting (emesis) is the forceful expulsion of gastric con- Allan, S.G. (1999) Nausea and vomiting. ogy of nausea and vomiting will promote a rational tents through the mouth (Twycross and Back, 1998). In: Doyle, D. et al (eds) Oxford Textbook choice of treatment. Nurses also need to be aware of After an episode of vomiting, there may be a post- of Palliative Medicine. Oxford: Oxford non-pharmacological measures that can reduce these ejection phase characterised by weakness, lethargy and University Press. distressing symptoms. shivering (Allan, 1999). Vomiting may ease the sensation Baines, M. (1997) Nausea, vomiting and of nausea. It may serve a protective function by expelling intestinal obstruction. -

Status of Brain Imaging in Gastroparesis
Review Status of Brain Imaging in Gastroparesis Zorisadday Gonzalez * and Richard W. McCallum Division of Gastroenterology, Center for Neurogastroenterology and GI Motility, Department of Internal Medicine, Texas Tech University Health Sciences Center, 4800 Alberta Ave., El Paso, TX 79905, USA; [email protected] * Correspondence: [email protected] Received: 6 March 2020; Accepted: 7 April 2020; Published: 9 April 2020 Abstract: The pathophysiology of nausea and vomiting in gastroparesis is complicated and multifaceted involving the collaboration of both the peripheral and central nervous systems. Most treatment strategies and studies performed in gastroparesis have focused largely on the peripheral effects of this disease, while our understanding of the central nervous system mechanisms of nausea in this entity is still evolving. The ability to view the brain with different neuroimaging techniques has enabled significant advances in our understanding of the central emetic reflex response. However, not enough studies have been performed to further explore the brain–gut mechanisms involved in nausea and vomiting in patients with gastroparesis. The purpose of this review article is to assess the current status of brain imaging and summarize the theories about our present understanding on the central mechanisms involved in nausea and vomiting (N/V) in patients with gastroparesis. Gaining a better understanding of the complex brain circuits involved in the pathogenesis of gastroparesis will allow for the development of better antiemetic prophylactic and treatment strategies. Keywords: gastroparesis; brain imaging; functional magnetic resonance imaging (fMRI); positron emission tomography (PET) scan; central nervous system (CNS) 1. Introduction Gastroparesis is a chronic heterogeneous motor disorder with variable clinical manifestations including episodic nausea, vomiting, retching, post-prandial fullness, early satiety, and/or upper abdominal pain in the absence of mechanical obstruction [1]. -

2019 Annual Report
2019 Annual Report [TITLE] Contents Centre Director’s Message 3 Objectives 4 IMPACCT at a glance 5 Awards and achievements 6 Research projects summary 7 Chronic Breathlessness management - World first medication listed 8 Publications summary 9 Postgraduate Palliative Care Course 10 IMPACCT Team 13 ImPaCCT:NSW Team 23 Clinical Trials 23 PaCCSC and CST Team, PaCCSC Governance 24 CST Governance 26 Consumer Advisory Group 30 External Academic Appointments 30 Editorial Roles 31 UTS Committees 32 UTS Teaching and Learning 33 IMPACCT-led Grants Awarded 2019 34 Collaborative grants led by other areas or institutions awarded in 2019 35 Current Projects 36 Higher degree research students 64 Publications 67 Conference presentations 77 Collaborations 78 ITCC Clinical Trials Sites 79 Visiting Scholars 83 External Engagement 84 Approvals & Overall Comments – Centre Annual Report 87 2019 ANNUAL REPORT | IMPACCT 2 Centre Director’s Message The Centre for Improving Palliative, Aged and Chronic Care through Clinical Research and Translation (IMPACCT) takes real world problems and works collaboratively to develop feasible, affordable and effective solutions. Combining the input of our lived-experience advisors (consumers), with the insights from our clinical experts, industry partners and the depth and breadth of expertise within our academic group, ensures that our research and educational endeavours are grounded in improving what matters most to older people - those living with progressive chronic illnesses and people with palliative care needs. This year, many of IMPACCT’s academics were honoured with a variety of awards, highlighted below and showcased in the Annual Report. One of the most significant achievements was the world’s first registration of a medication to manage optimally treated breathlessness, and its subsequent addition to the pharmaceutical benefits scheme list in early 2019 was a major highlight. -

A 58-Year-Old Man with Esophageal Cancer and Nausea, Vomiting, and Intractable Hiccups
The new england journal of medicine case records of the massachusetts general hospital Founded by Richard C. Cabot Nancy Lee Harris, m.d., Editor Jo-Anne O. Shepard, m.d., Associate Editor Stacey M. Ellender, Assistant Editor Sally H. Ebeling, Assistant Editor Christine C. Peters, Assistant Editor Case 6-2005: A 58-Year-Old Man with Esophageal Cancer and Nausea, Vomiting, and Intractable Hiccups Eric L. Krakauer, M.D., Ph.D., Andrew X. Zhu, M.D., Ph.D., Brenna C. Bounds, M.D., Dushyant Sahani, M.D., Kevin R. McDonald, M.D., and Elena F. Brachtel, M.D. presentation of case Dr. Kevin R. McDonald: A 58-year-old man was transferred to this hospital for manage- From the Palliative Care Service (E.L.K., ment of newly diagnosed adenocarcinoma of the esophagus. K.R.M.), the Division of Hematology and Oncology (A.X.Z.), the Gastroenterology Several months before admission, the patient initially had episodes of early satiety Unit (B.C.B.), and the Departments of Ra- and then began to vomit all solid food. He lost 14 kg; light-headedness and dyspnea with diology (D.S.) and Pathology (E.F.B.), minimal exertion developed. He did not have nausea, dysphagia, or odynophagia, but Massachusetts General Hospital; and the Departments of Medicine (E.L.K., A.X.Z., he was able to ingest only small amounts of liquids slowly without vomiting. Twelve B.C.B., K.R.M.), Radiology (D.S.), and Pa- days before admission, he went to the emergency department of another hospital. thology (E.F.B.), Harvard Medical School. -

Nauseated and Breathless: Nobody’S Preferred Way to Die Annie Massart, MD [email protected] Twitter: @Annie Massart Disclosures
Nauseated and Breathless: Nobody’s Preferred Way to Die Annie Massart, MD [email protected] Twitter: @Annie_Massart_ Disclosures • None Objectives • Describe the prevalence of dyspnea in advanced COPD and lung cancer • Identify therapeutic options for managing dyspnea • Name the mechanisms of action for common antiemetics • Tailor antiemetic regimens to nausea triggers Case 1 • 37.6, HR 95, BP 130/80, RR 20, O2 sat 94% on RA. • Normal work of breathing, decreased air movement throughout, no wheezes. While speaking, some pursed lip breathing and has to pause intermittently to breathe. • Normal bowel sounds, LLQ TTP, no rebound/guarding • CT abdomen: uncomplicated diverticulitis • CXR: stable emphysema, no infiltrate Case 1 How would you address his breathing concerns? A. Start supplemental oxygen and monitor response B. Treat for COPD exacerbation with steroids and nebulized bronchodilators C. Start him on Morphine Extended Release 30 mg po q12h and monitor response D. Provide him with a hand-held fan and monitor response E. Remind him he’s here for diverticulitis and you’ll refer him to pulmonary clinic Kamal, A.H., et al. J Palliat Med. 2011 Oct;14(10):1167-72 Dyspnea • 50-80% of patients with advanced cancer • 90-95% of patients with advanced COPD • Dyspnea and distress both higher in COPD than lung cancer Dyspnea • Decreased activity level • Deconditioned patients become dyspneic more easily • Increased dependence on their caregivers • Decreased quality of life Assessing Dyspnea O’Donnell, D.E., et al. Can Respir J. 2007 Sep;14 Suppl -
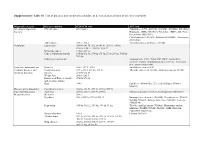
Supplementary Table S1. List of Diseases and Conditions Candidate to Be Tested As Predictors of One-Year Mortality
Supplementary Table S1. List of diseases and conditions candidate to be tested as predictors of one-year mortality Diagnostic category Disease/condition ICD-9 CM code ATC code Infectious and parasitic HIV infection 042.x-044.x Zidovudine (AZT) (J05AF01, J05AR01, J05AR04, J05AR05), diseases Didanosine (DDI) (J05AF02), Zalcitabine (DDC) (J05AF03), Pentamidine (P01CX01), Clarithromycin (J01FA09), Rifabutin (J04AB04), Atovaquone (P01AX06) Tuberculosis 010.x - 018.x Anti-tuberculosis antibiotics (J04AB) Neoplasms Lymphoma 200.00-202.38, 202.50-203.01, 203.8x, 238.6x, 273.3x, V10.71, V10.72, V10.79 Metastatic cancer 196.0x-199.1x Cancer, without metastasis 140.0x-172.9x, 174.0x-175.9x,179.x-195.8x, V10.0x- V10.9x Malignancy medication Antineoplastic (L01), Taxol (C07AB05), Interleukins (L03AC), Colony-stimulating factors (L03AA), Antinausea misc, ondansetron (A04) Endocrine, nutritional and Diabetes 250.x, 357.2, 362.0 Antidiabetic agents (A10) metabolic diseases, and Hypothyroidism 243.x-244.2, 244.8x, 244.9x Thyroid replacement (H03A), Antithyroid agents (H03B) immunity disorders Obesity 278.00-278.01 Weight loss 260.0x-263.9 Disorders of fluid, electrolyte, 276.0x-276.9x and acid-base balance Gout 274.x Colchicine (M04AC01), Uric acid inhibitors (M04AA, M04AB) Diseases of the blood and Coagulation defects 286.0x-286.9x, 287.1x, 287.3x-287.5x blood-forming organs Anaemias 280.0x, 280.1x-281.9x, 285.9x Marrow stimulants (L03AA), Erythropoietin (B03XA01) Mental disorders Dementia 290.x Psychosis 295.x-298.9x, 299.10-299.11 Butyrophenone derivates -

A Multicenter Phase I Study Evaluating Dual PI3K and BRAF
Published OnlineFirst October 19, 2017; DOI: 10.1158/1078-0432.CCR-17-1807 Cancer Therapy: Clinical Clinical Cancer Research A Multicenter Phase I Study Evaluating Dual PI3K and BRAF Inhibition with PX-866 and Vemurafenib in Patients with Advanced BRAF V600–Mutant Solid Tumors Clinton Yam1,2, Xiaowei Xu1, Michael A. Davies2, Phyllis A. Gimotty3, Jennifer J.D. Morrissette4, Michael T. Tetzlaff2, Khalida M. Wani2, Shujing Liu1, Wanleng Deng2, Meghan Buckley3, Jianhua Zhao4, Ravi K. Amaravadi1, Naomi B. Haas1, Ragini R. Kudchadkar5, Anna C. Pavlick6, Jeffrey A. Sosman7, Hussein Tawbi2,8, Luke Walker9, Lynn M. Schuchter1, Giorgos C. Karakousis1, and Tara C. Gangadhar1 Abstract Purpose: The objectives of the study were to evaluate the safety 3 (PX-866 8 mg daily; vemurafenib 960 mg twice daily), respec- of daily oral PX-866 in combination with twice daily vemurafenib tively. Of 23 response-evaluable patients, seven had confirmed and to identify potential predictive biomarkers for this novel partial responses (PR), 10 had stable disease, and six had disease combination. progression. Decreases in intratumoral pAKT expression were Experimental Design: We conducted a phase I, open-label, observed following treatment with PX-866. Patients who dose-escalation study in patients with advanced BRAF V600– achieved PRs had higher rates of PTEN loss by IHC (80% vs. mutant solid tumors. PX-866 was administered on a continuous 58%) and pathogenic PTEN mutations and/or deletions (57% vs. schedule in combination with vemurafenib. Patients underwent a 25%). Two patients with durable PRs had an increase in intratu- þ baseline and on-treatment biopsy after 1-week of PX-866 mono- moral CD8 T-cell infiltration following treatment with PX-866. -

Berkshire Adult Palliative Care Guidelines Best Practice Document
Berkshire West Integrated Care System Representing Berkshire West Clinical Commisioning Group Royal Berkshire NHS Foundation Trust Berkshire Healthcare NHS Foundation Trust Berkshire West Primary Care Alliance Berkshire Adult Palliative Care Guidelines Best Practice Document [APC ClinDoc 016a] F For the latest information on interactions and adverse effects, always consult the latest version of the Summary of Product Characteristics (SPC), which can be found at: http://www.medicines.org.uk/ A Approval and Authorisation Approved by Job Title Date Area Prescribing APC Chair November 2019 Committee C Change History Version Date Author Reason v.2.0 01/10/2018 Specialist Palliative Care Medical Updated APC Category Teams East Berkshire (Thames Hospice/HWPH) and West Berkshire (Sue Ryder Duchess of Kent and Nettlebed and RBHFT) This prescribing guideline remains open to review considering any new evidence This guideline should only be viewed online and will no longer be valid if printed off or saved locally POLICY DEVELOPMENT ADULT PALLIATIVE CARE GUIDELINES SECTION 1: PAIN – BEST PRACTICE DOCUMENT SECTION 2: OTHER SYMPTOMS – BEST PRACTICE DOCUMENT SECTION 2: OTHER SYMPTOMS – BEST PRACTICE DOCUMENT History These guidelines cover all services in Berkshire including Wexham Park Hospital, Royal Berks Hospital, BHFT, East Berkshire CCGs, Sue Ryder Hospices & Thames Hospice. They are to be read in conjunction with Organisational Policies on Medicines Management, Prescribing and administration of medicines and use of Oxygen appropriate to each organization. -

Extended S3 Guideline Palliative Care for Patients with Incurable Cancer
published by: 1.1 Editors Extended S3 Guideline Palliative care for patients with incurable cancer Short version 2.2 – September 2020 AWMF- Registration number: 128/001OL 2 Important New Features in the Ex- tended 2019 Version of the Guideline A. Eight new chapters have been written as part of the second development phase of the guideline (2016-2019) and added to the seven original chapters: • Defining Goals of Care and Criteria for Clinical Decision-making • Fatigue • Sleep-related Illnesses/Nocturnal Restlessness • Nausea and Vomiting (not Tumour Therapy-related) • Malignant Bowel Obstruction (MBO) • Malignant Wounds • Anxiety • The Desire to Die B. In the seven chapters written during the first development phase in 2011-2015, all recommendations were checked to ensure that they were up-to-date, and updated if necessary (chapters on organisation of palliative care, communication, breathlessness, cancer pain, constipation, depression, dying phase). For this purpose, a systematic search for prioritised topics and a survey of the experts involved was conducted. As part of the updating process, the recommendations listed below have been revised. In some recommendations, only the level of evidence was changed on the basis of new study data (marked with a #). • Recommendations 4.4., 4.5., 4.6. and 4.7. (Principles of Palliative Care) • Recommendations 5.1.# and 5.2.# (Organisation of Palliative Care - Time of Palliative Care Integration) • Recommendations 5.8.# and 5.9.# (Organisation of Palliative Care - Assessment of Patient Needs and Determining Complexity) • Recommendation 8.2. (Breathlessness - Assessment) • Recommendation 8.17. (Breathlessness - Steroids (Glucocorticoids)) • Recommendation 9.5.# (Cancer pain - WHO Step III First-choice Opioids) • Recommendation 9.28 (Cancer pain - Treatment of Opioid-related Constipation) • Recommendation 13.1. -

Medical Classifications
ANNEX F ANNEX F Medical Classifications A SYSTEM OF HEALTH ACCOUNTS 2011: REVISED EDITION © OECD, EUROPEAN UNION, WORLD HEALTH ORGANIZATION 2017 497 ANNEX F Table F.1.1. International Classification of Primary Care (ICPC-2) ICPC-2 – English Blood, Blood Forming Eye F Musculoskeletal L International Classification of F01 Eye pain L01 Neck symptom/complain Organs and Immune L02 Back symptom/complaint Primary Care – 2 nd Edition F02 Red eye Mechanism B F03 Eye discharge L03 Low back symptom/complaint F04 Visual floaters/spots L04 Chest symptom/complaint Wonca International B02 Lymph gland(s) enlarged/painful F05 Visual disturbance other L05 Flank/axilla symptom/complaint B04 Blood symptom/complaint Classification Committee F13 Eye sensation abnormal L07 Jaw symptom/complaint B25 Fear of aids/HIV F14 Eye movements abnormal L08 Shoulder symptom/complaint (WICC) B26 Fear cancer blood/lymph F15 Eye appearance abnormal L09 Arm symptom/complaint B27 Fear blood/lymph disease other F16 Eyelid symptom/complaint L10 Elbow symptom/complaint B28 Limited function/disability F17 Glasses symptom/complaint L11 Wrist symptom/complaint B29 Sympt/ complt lymph/immune other Process codes F18 Contact lens symptom/complaint L12 Hand/finger symptom/complaint B70 Lymphadenitis acute -30 Medical Exam/ Eval-Complete F27 Fear of eye disease L13 Hip symptom/complaint B71 Lymphadenitis non-specific -31 Medical Examination/Health Evaluation- F28 Limited function/disability (f) L14 Leg/thigh symptom/complaint B72 Hodgkin's disease/lymphoma Partial/Pre-op check F29 Eye