Role of Rho-Family Guanosine Triphosphatase Effectors in Filopodia Dynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Anti-Integrin Beta 5 PE Catalog Number: 12-0497 Also Known As:Integrin B5, ITGB5 RUO: for Research Use Only
Anti-Integrin beta 5 PE Catalog Number: 12-0497 Also Known As:Integrin b5, ITGB5 RUO: For Research Use Only. Not for use in diagnostic procedures. Staining of CHO cells with Mouse IgG1 kappa Isotype Control PE (cat. 12-4714) (open histogram) or Anti-Integrin beta 5 PE (filled histogram). Total viable cells were used for analysis. Product Information Contents: Anti-Integrin beta 5 PE Formulation: aqueous buffer, 0.09% sodium azide, may Catalog Number: 12-0497 contain carrier protein/stabilizer Clone: KN52 Temperature Limitation: Store at 2-8°C. Do not freeze. Concentration: 5 uL (0.06 ug)/test Light sensitive material. Host/Isotype: Mouse IgG1, kappa Batch Code: Refer to Vial Use By: Refer to Vial Caution, contains Azide Description The KN52 antibody reacts with human integrin beta 5, an approximately 100,000 Mr (reduced) and 95,000 Mr (nonreduced) a member of the integrin family. Only a single alpha chain, the alpha v subunit, associates with the integrin beta 5 to form the vitronectin receptor complex. Integrin alpha v/beta 5 complex also binds to the basic domain of Tat (he sequence RKKRRQRRR). The integrin beta 5 is found on carcinoma cell lines, hepatoma and fibroblast cell lines, and is absent from lymphoblastoid cells and platelets. The KN52 monoclonal antibody has been shown to inhibit migration. Applications Reported This KN52 antibody has been reported for use in flow cytometric analysis. Applications Tested This KN52 antibody has been pre-titrated and tested by flow cytometric analysis of Chinese Hamster Ovary (CHO), A549 and normal human peripheral blood cells. -

RT² Profiler PCR Array (Rotor-Gene® Format) Mouse Focal Adhesions
RT² Profiler PCR Array (Rotor-Gene® Format) Mouse Focal Adhesions Cat. no. 330231 PAMM-145ZR For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Rotor-Gene Q, other Rotor-Gene cyclers Format R Description The Mouse Focal Adhesions RT² Profiler PCR Array profiles the expression of 84 key genes involved in cellular adhesion to the extracellular matrix (ECM). Focal adhesions and hemidesmosomes form around the intracellular domains of integrins bound to components of the ECM such as those found in the basal lamina underlying epithelial cells. Focal adhesions connect the intracellular domains of integrins to actin filaments, while hemidesmosomes connect them to keratin-based intermediate filaments. These structures regulate several key normal biological processes including angiogenesis, anchorage-dependent cell survival, cell cycle, cell migration, and wound healing. Dysregulation of focal adhesion function and integrity plays a key role in the pathophysiology of diseases such as fibrosis and epithelial-to-mesenchymal transition during tumor metastasis. The well-studied focal adhesion kinase PTK2, a cytosolic tyrosine kinase, and integrin-linked kinase (ILK) mediate PI-3-kinase/AKT and G-protein integrin-dependent signaling associated with focal adhesion-dependent processes. Downstream signaling from focal adhesions and caveolae, as well as focal-adhesion–interacting proteins (such as filamin, vinculin, and talin), then regulate the biogenesis, organization, polymerization, and depolymerization of actin filaments. Profiling the expression of focal adhesion and hemidesmosome components may lead to a better understanding of molecular mechanisms behind cell-ECM contact mediated cell biology. Using real-time PCR, research studies can easily and reliably analyze the expression of a focused panel of genes involved in focal adhesions with this array. -

Integrin and Gene Network Analysis Reveals That ITGA5 and ITGB1 Are Prognostic in Non-Small-Cell Lung Cancer
Journal name: OncoTargets and Therapy Article Designation: Original Research Year: 2016 Volume: 9 OncoTargets and Therapy Dovepress Running head verso: Zheng et al Running head recto: ITGA5 and ITGB1 are prognostic in NSCLC open access to scientific and medical research DOI: http://dx.doi.org/10.2147/OTT.S91796 Open Access Full Text Article ORIGINAL RESEARCH Integrin and gene network analysis reveals that ITGA5 and ITGB1 are prognostic in non-small-cell lung cancer Weiqi Zheng Background: Integrin expression has been identified as a prognostic factor in non-small-cell Caihui Jiang lung cancer (NSCLC). This study was aimed at determining the predictive ability of integrins Ruifeng Li and associated genes identified within the molecular network. Patients and methods: A total of 959 patients with NSCLC from The Cancer Genome Atlas Department of Radiation Oncology, Guangqian Hospital, Quanzhou, Fujian, cohorts were enrolled in this study. The expression profile of integrins and related genes were People’s Republic of China obtained from The Cancer Genome Atlas RNAseq database. Clinicopathological characteristics, including age, sex, smoking history, stage, histological subtype, neoadjuvant therapy, radiation therapy, and overall survival (OS), were collected. Cox proportional hazards regression models as well as Kaplan–Meier curves were used to assess the relative factors. Results: In the univariate Cox regression model, ITGA1, ITGA5, ITGA6, ITGB1, ITGB4, and ITGA11 were predictive of NSCLC prognosis. After adjusting for clinical factors, ITGA5 (odds ratio =1.17, 95% confidence interval: 1.05–1.31) andITGB1 (odds ratio =1.31, 95% confidence interval: 1.10–1.55) remained statistically significant. In the gene cluster network analysis, PLAUR, ILK, SPP1, PXN, and CD9, all associated with ITGA5 and ITGB1, were identified as independent predictive factors of OS in NSCLC. -

Eosinophil Accumulation in Postnatal Lung Is Specific to the Primary
www.nature.com/scientificreports OPEN Eosinophil accumulation in postnatal lung is specifc to the primary septation phase of development Lucas F. Lofredo1,4, Mackenzie E. Coden1,4, Brian M. Jeong1, Matthew T. Walker1, Kishore Reddy Anekalla 2, Ton C. Doan1, Raul Rodriguez1, Mandy Browning1, Kiwon Nam1, James J. Lee3, Hiam Abdala-Valencia2 & Sergejs Berdnikovs1* Type 2 immune cells and eosinophils are transiently present in the lung tissue not only in pathology (allergic disease, parasite expulsion) but also during normal postnatal development. However, the lung developmental processes underlying airway recruitment of eosinophils after birth remain unexplored. We determined that in mice, mature eosinophils are transiently recruited to the lung during postnatal days 3–14, which specifcally corresponds to the primary septation/alveolarization phase of lung development. Developmental eosinophils peaked during P10-14 and exhibited Siglec-Fmed/highCD11c−/low phenotypes, similar to allergic asthma models. By interrogating the lung transcriptome and proteome during peak eosinophil recruitment in postnatal development, we identifed markers that functionally capture the establishment of the mesenchymal-epithelial interface (Nes, Smo, Wnt5a, Nog) and the deposition of the provisional extracellular matrix (ECM) (Tnc, Postn, Spon2, Thbs2) as a key lung morphogenetic event associating with eosinophils. Tenascin-C (TNC) was identifed as one of the key ECM markers in the lung epithelial-mesenchymal interface both at the RNA and protein levels, consistently associating with eosinophils in development and disease in mice and humans. As determined by RNA-seq analysis, naïve murine eosinophils cultured with ECM enriched in TNC signifcantly induced expression of Siglec-F, CD11c, eosinophil peroxidase, and other markers typical for activated eosinophils in development and allergic infammatory responses. -

CD81 Controls Beige Fat Progenitor Cell Growth and Energy Balance Via FAK Signaling
UCSF UC San Francisco Previously Published Works Title CD81 Controls Beige Fat Progenitor Cell Growth and Energy Balance via FAK Signaling. Permalink https://escholarship.org/uc/item/1fb2d4gg Journal Cell, 182(3) ISSN 0092-8674 Authors Oguri, Yasuo Shinoda, Kosaku Kim, Hyeonwoo et al. Publication Date 2020-08-01 DOI 10.1016/j.cell.2020.06.021 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Article CD81 Controls Beige Fat Progenitor Cell Growth and Energy Balance via FAK Signaling Graphical Abstract Authors Yasuo Oguri, Kosaku Shinoda, Hyeonwoo Kim, ..., Suneil K. Koliwad, Bruce M. Spiegelman, Shingo Kajimura Correspondence [email protected] In Brief A subset of adipocyte progenitor cells give rise to beige fat through signaling responses to irisin through the action of specific integrins and the co- receptor CD81. Highlights d Beige fat progenitors are marked by cell surface proteins, PDGFRa, Sca1, and CD81 d Beige APC proliferation is regulated by temperature, genetic background, and aging d CD81 mediates integrin-FAK signaling in response to irisin d CD81 loss causes obesity, insulin resistance, and adipose tissue inflammation Oguri et al., 2020, Cell 182, 1–15 August 6, 2020 ª 2020 Elsevier Inc. https://doi.org/10.1016/j.cell.2020.06.021 ll Please cite this article in press as: Oguri et al., CD81 Controls Beige Fat Progenitor Cell Growth and Energy Balance via FAK Signaling, Cell (2020), https://doi.org/10.1016/j.cell.2020.06.021 ll Article CD81 Controls Beige Fat Progenitor Cell Growth and Energy Balance via FAK Signaling Yasuo Oguri,1,2,3,4,13 Kosaku Shinoda,1,2,3,5,13 Hyeonwoo Kim,6,13 Diana L. -
![RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)](https://docslib.b-cdn.net/cover/3271/rt%C2%B2-profiler-pcr-array-96-well-format-and-384-well-4-x-96-format-3353271.webp)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 X 96] Format)
RT² Profiler PCR Array (96-Well Format and 384-Well [4 x 96] Format) Mouse Focal Adhesions Cat. no. 330231 PAMM-145ZA For pathway expression analysis Format For use with the following real-time cyclers RT² Profiler PCR Array, Applied Biosystems® models 5700, 7000, 7300, 7500, Format A 7700, 7900HT, ViiA™ 7 (96-well block); Bio-Rad® models iCycler®, iQ™5, MyiQ™, MyiQ2; Bio-Rad/MJ Research Chromo4™; Eppendorf® Mastercycler® ep realplex models 2, 2s, 4, 4s; Stratagene® models Mx3005P®, Mx3000P®; Takara TP-800 RT² Profiler PCR Array, Applied Biosystems models 7500 (Fast block), 7900HT (Fast Format C block), StepOnePlus™, ViiA 7 (Fast block) RT² Profiler PCR Array, Bio-Rad CFX96™; Bio-Rad/MJ Research models DNA Format D Engine Opticon®, DNA Engine Opticon 2; Stratagene Mx4000® RT² Profiler PCR Array, Applied Biosystems models 7900HT (384-well block), ViiA 7 Format E (384-well block); Bio-Rad CFX384™ RT² Profiler PCR Array, Roche® LightCycler® 480 (96-well block) Format F RT² Profiler PCR Array, Roche LightCycler 480 (384-well block) Format G RT² Profiler PCR Array, Fluidigm® BioMark™ Format H Sample & Assay Technologies Description The Mouse Focal Adhesions RT² Profiler PCR Array profiles the expression of 84 key genes involved in cellular adhesion to the extracellular matrix (ECM). Focal adhesions and hemidesmosomes form around the intracellular domains of integrins bound to components of the ECM such as those found in the basal lamina underlying epithelial cells. Focal adhesions connect the intracellular domains of integrins to actin filaments, while hemidesmosomes connect them to keratin-based intermediate filaments. These structures regulate several key normal biological processes including angiogenesis, anchorage-dependent cell survival, cell cycle, cell migration, and wound healing. -

Probe Set Name Symbol 1598 G at Growth Arres
Supplementary Table 2. List of stroma related genes (i.e. probe sets overexpressed in core relative to FNA biopsies of the same cancer) Probe set Name Symbol 1598_g_at growth arrest-specific 6 GAS6 200048_s_at jumping translocation breakpoint JTB 200054_at zinc finger protein 259 ZNF259 200055_at TAF10 RNA polymerase II, TATA box binding protein (TBP)-associatedTAF10 factor, 30kDa 200059_s_at ras homolog gene family, member A RHOA 200060_s_at RNA binding protein S1, serine-rich domain RNPS1 200070_at chromosome 2 open reading frame 24 C2orf24 200613_at adaptor-related protein complex 2, mu 1 subunit AP2M1 200663_at CD63 molecule CD63 200665_s_at secreted protein, acidic, cysteine-rich (osteonectin) SPARC 200671_s_at spectrin, beta, non-erythrocytic 1 SPTBN1 200696_s_at gelsolin (amyloidosis, Finnish type) GSN 200704_at lipopolysaccharide-induced TNF factor LITAF 200738_s_at phosphoglycerate kinase 1 PGK1 200760_s_at ADP-ribosylation-like factor 6 interacting protein 5 ARL6IP5 200762_at dihydropyrimidinase-like 2 DPYSL2 200770_s_at laminin, gamma 1 (formerly LAMB2) LAMC1 200771_at laminin, gamma 1 (formerly LAMB2) LAMC1 200772_x_at prothymosin, alpha PTMA 200778_s_at septin 2 2-Sep 200782_at annexin A5 ANXA5 200784_s_at low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor)LRP1 200785_s_at low density lipoprotein-related protein 1 (alpha-2-macroglobulin receptor)LRP1 200795_at SPARC-like 1 (hevin) SPARCL1 200799_at heat shock 70kDa protein 1A HSPA1A 200807_s_at heat shock 60kDa protein 1 (chaperonin) HSPD1 200811_at -

IPI00420067 ADAM Metallope
Supplementary material Ann Rheum Dis ID Gene Name gene name main functions assosiation with oc\ob\cartilage\disc(np af ep notochord cells) IPI00420067 ADAM metallopeptidase domain 15 IPI00288894 ADAM metallopeptidase domain 17 IPI00307592 ATP-binding cassette, sub-family A (ABC1), member 2 IPI00291373 ATP-binding cassette, sub-family D (ALD), member 1 IPI00002372 ATP-binding cassette, sub-family D (ALD), member 3 IPI00784119 ATPase, H+ transporting, lysosomal accessory protein 1 IPI00021391 C-type lectin domain family 2, member B IPI00788676 CD109 molecule IPI00384548 CD63 molecule IPI00736241 CUB domain containing protein 1 IPI00844115 DnaJ (Hsp40) homolog, subfamily C, member 10 IPI00005683 ER degradation enhancer, mannosidase alpha-like 1 IPI00892978 ER degradation enhancer, mannosidase alpha-like 3 IPI00007940 ER lipid raft associated 1 IPI00026942 ER lipid raft associated 2 IPI00383663 ERGIC and golgi 3 IPI00872773 ERO1-like (S. cerevisiae) IPI00334818 FK506 binding protein 10, 65 kDa IPI00943593 Fraser syndrome 1 IPI00879033 G protein-coupled receptor 110 IPI00445699 KDEL (Lys-Asp-Glu-Leu) containing 2 IPI00645487 KIAA0090 IPI00885086 KIAA0746 protein IPI00456649 KIAA1161 IPI00008787 N-acetylglucosaminidase, alpha- IPI00940046 N-acylsphingosine amidohydrolase (acid ceramidase) 1 IPI00005600 N-deacetylase/N-sulfotransferase (heparan glucosaminyl) 2 IPI00019988 N-sulfoglucosamine sulfohydrolase IPI00794861 NEL-like 2 (chicken) Wang G, et al. Ann Rheum Dis 2020;0:1–10. doi: 10.1136/annrheumdis-2019-216911 Supplementary material Ann -

Large-Scale Gene Expression Analysis of Osteoblasts Cultured on Three Different Ti–6Al–4V Surface Treatments Ching-Hsin Kua,B, Martin Browneb, Peter J
CORE Metadata, citation and similar papers at core.ac.uk Provided by Infoscience - École polytechnique fédérale de Lausanne Biomaterials 23 (2002) 4193–4202 Large-scale gene expression analysis of osteoblasts cultured on three different Ti–6Al–4V surface treatments Ching-Hsin Kua,b, Martin Browneb, Peter J. Gregsonb, Jacques Corbeild, Dominique P. Piolettia,c,* a Bone Bioengineering Group, Institute for Biomedical Engineering, Swiss Federal Institute of Technology, Lausanne, Switzerland b Bioengineering Sciences Research Group, School of Engineering Sciences, University of Southampton, Southampton, UK c Orthopaedic Hospital, Lausanne, Switzerland d Department of Medicine, University of California, San Diego and San Diego Veterans Affairs, Healthcare System, La Jolla, USA Received 24 October 2001 Abstract To improve implant biocompatibility, we developed a simple cost-effective thermal surface treatment allowing an increase in the oxide layer thickness of a titanium (Ti) alloy used in orthopaedic implants. The goal of this study was to test in vitro the reaction of osteoblasts to the developed surface treatment and to compare it to the osteoblast reaction to two other surface treatments currently used in the practice of implant surgery. Quantification of osteoblast gene expression on a large scale was used in this study. The kinetics of gene expression over 120 h was followed for 58 genes to quantify the effect of the developed surface treatment. Twenty eight genes were further selected to compare the effects of surface treatments on osteoblasts. Based on the genes studied, we could propose a general pathway for the cell reaction according to the surface treatments used: (1) metal ion release changes the time course of gene expression in the FAK pathway; (2) once the accumulation of metal ions released from the Ti surface exceeds a threshold value, cell growth is diminished and apoptosis may be activated; (3) PTK up-regulation is also induced by metal ion release; (4) the expression of Bcl-2 family and Bax may suggest that metal ions induce apoptosis. -

Cell Adhesion Molecules Are Mediated by Photobiomodulation at 660 Nm in Diabetic Wounded Fibroblast Cells
cells Article Cell Adhesion Molecules Are Mediated by Photobiomodulation at 660 nm in Diabetic Wounded Fibroblast Cells Nicolette N. Houreld * ID , Sandra M. Ayuk and Heidi Abrahamse ID Laser Research Centre, Faculty of Health Sciences, University of Johannesburg, P.O. Box 17011, Doornfontein, Johannesburg 2028, South Africa; [email protected] (S.M.A.); [email protected] (H.A.) * Correspondence: [email protected]; Tel.: +27-11-559-6833 Received: 9 March 2018; Accepted: 12 April 2018; Published: 16 April 2018 Abstract: Diabetes affects extracellular matrix (ECM) metabolism, contributing to delayed wound healing and lower limb amputation. Application of light (photobiomodulation, PBM) has been shown to improve wound healing. This study aimed to evaluate the influence of PBM on cell adhesion molecules (CAMs) in diabetic wound healing. Isolated human skin fibroblasts were grouped into a diabetic wounded model. A diode laser at 660 nm with a fluence of 5 J/cm2 was used for irradiation and cells were analysed 48 h post-irradiation. Controls consisted of sham-irradiated (0 J/cm2) cells. Real-time reverse transcription (RT) quantitative polymerase chain reaction (qPCR) was used to determine the expression of CAM-related genes. Ten genes were up-regulated in diabetic wounded cells, while 25 genes were down-regulated. Genes were related to transmembrane molecules, cell–cell adhesion, and cell–matrix adhesion, and also included genes related to other CAM molecules. PBM at 660 nm modulated gene expression of various CAMs contributing to the increased healing seen in clinical practice. There is a need for new therapies to improve diabetic wound healing. -
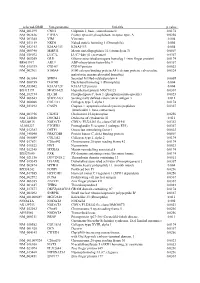
Supplementary Figures S1-S3
selected-GBID Uni-genename Uni-title p value NM_001299 CNN1 Calponin 1, basic, smooth muscle 0.0174 NM_002836 PTPRA Protein tyrosine phosphatase, receptor type, A 0.0256 NM_003380 VIM Vimentin 0.004 NM_033119 NKD1 Naked cuticle homolog 1 (Drosophila) 0.004 NM_052913 KIAA1913 KIAA1913 0.004 NM_005940 MMP11 Matrix metallopeptidase 11 (stromelysin 3) 0.0069 NM_018032 LUC7L LUC7-like (S. cerevisiae) 0.0367 NM_005269 GLI1 Glioma-associated oncogene homolog 1 (zinc finger protein) 0.0174 BE463997 ARL9 ADP-ribosylation factor-like 9 0.0367 NM_015939 CGI-09 CGI-09 protein 0.0023 NM_002961 S100A4 S100 calcium binding protein A4 (calcium protein, calvasculin, 0.0324 metastasin, murine placental homolog) NM_003014 SFRP4 Secreted frizzled-related protein 4 0.0005 NM_080759 DACH1 Dachshund homolog 1 (Drosophila) 0.004 NM_053042 KIAA1729 KIAA1729 protein 0.004 BX415194 MGC16121 Hypothetical protein MGC16121 0.0367 NM_182734 PLCB1 Phospholipase C, beta 1 (phosphoinositide-specific) 0.0023 NM_006643 SDCCAG3 Serologically defined colon cancer antigen 3 0.011 NM_000088 COL1A1 Collagen, type I, alpha 1 0.0174 NM_033292 CASP1 Caspase 1, apoptosis-related cysteine peptidase 0.0367 (interleukin 1, beta, convertase) NM_003956 CH25H Cholesterol 25-hydroxylase 0.0256 NM_144658 DOCK11 Dedicator of cytokinesis 11 0.011 AK024935 NODATA CDNA: FLJ21283 fis, clone COL01910 0.0363 AL050227 PTGER3 Prostaglandin E receptor 3 (subtype EP3) 0.0367 NM_012383 OSTF1 Osteoclast stimulating factor 1 0.0023 NM_145040 PRKCDBP Protein kinase C, delta binding protein 0.0069 NM_000089