Real-Time Quantitative Reverse Transcriptase-Polymerase Chain Reaction Analysis of Melanoma Progression-Associated Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Functions of the Mineralocorticoid Receptor in the Hippocampus By
Functions of the Mineralocorticoid Receptor in the Hippocampus by Aaron M. Rozeboom A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cellular and Molecular Biology) in The University of Michigan 2008 Doctoral Committee: Professor Audrey F. Seasholtz, Chair Professor Elizabeth A. Young Professor Ronald Jay Koenig Associate Professor Gary D. Hammer Assistant Professor Jorge A. Iniguez-Lluhi Acknowledgements There are more people than I can possibly name here that I need to thank who have helped me throughout the process of writing this thesis. The first and foremost person on this list is my mentor, Audrey Seasholtz. Between working in her laboratory as a research assistant and continuing my training as a graduate student, I spent 9 years in Audrey’s laboratory and it would be no exaggeration to say that almost everything I have learned regarding scientific research has come from her. Audrey’s boundless enthusiasm, great patience, and eager desire to teach students has made my time in her laboratory a richly rewarding experience. I cannot speak of Audrey’s laboratory without also including all the past and present members, many of whom were/are not just lab-mates but also good friends. I also need to thank all the members of my committee, an amazing group of people whose scientific prowess combined with their open-mindedness allowed me to explore a wide variety of interests while maintaining intense scientific rigor. Outside of Audrey’s laboratory, there have been many people in Ann Arbor without whom I would most assuredly have gone crazy. -

Methylation and Silencing of Protein Tyrosine Phosphatase Receptor
Human Cancer Biology Methylation and Silencing of Protein Tyrosine Phosphatase Receptor Type O in Chronic Lymphocytic Leukemia Tasneem Motiwala,1Sarmila Majumder,1Huban Kutay,1David Spencer Smith,1Donna S. Neuberg,4 David M. Lucas,2 John C. Byrd,2,3 Michael Grever,2,3 and Samson T.Jacob1, 2 , 3 Abstract Purpose: Previous studies in our laboratory have shown the progressive methylation and suppression of the gene encoding protein tyrosine phosphatase, PTPRO, in the livers of rats fed a methyl-deficient diet that induces hepatocarcinogenesis. Subsequently, we observed the methylation of PTPRO in primary human lung tumors and also showed its potential tumor suppressor characteristics. The present study was undertaken to investigate whether the truncated form of PTPRO (PTPROt), specifically expressed in naI«ve B lymphocytes, was also methylated and suppressed in chronic lymphocytic leukemia (CLL), a disease generally affecting B lymphocytes. Experimental Design and Results: Initial screening showed that 60% of the 52 CLL samples analyzed using methylation-specific PCR assay were methylated compared with B lymphocytes from normal individuals, which were not methylated. The expression of PTPROt, as measured by semiquantitative reverse transcription-PCR, inversely correlated with methylation in the few samples tested. Analysis of additional samples (n = 50) by combined bisulfite restriction analysis showed that the PTPRO CpG island was methylated in 82% of patients with CLL compared with B lymphocytes from normal individuals. Furthermore, overall expression of PTPRO was reduced in CLL relative to normal lymphocytes. The PTPRO gene was also suppressed by methylation in the CLL cell lineWaC3CD5, where it could be reactivated upon treatment with the DNA hypome- thylating agent 5-AzaC. -

Identification of Chebulinic Acid As a Dual Targeting Inhibitor of Protein
Bioorganic Chemistry 90 (2019) 103087 Contents lists available at ScienceDirect Bioorganic Chemistry journal homepage: www.elsevier.com/locate/bioorg Short communication Identification of chebulinic acid as a dual targeting inhibitor of protein T tyrosine phosphatases relevant to insulin resistance Sun-Young Yoona,1, Hyo Jin Kangb,1, Dohee Ahna, Ji Young Hwanga, Se Jeong Kwona, ⁎ Sang J. Chunga, a School of Pharmacy, Sungkyunkwan University, Suwon 16419, Republic of Korea b Department of Chemistry, Dongguk University, Seoul 100-715, Republic of Korea ARTICLE INFO ABSTRACT Keywords: Natural products as antidiabetic agents have been shown to stimulate insulin signaling via the inhibition of the Protein tyrosine phosphatases (PTPs) protein tyrosine phosphatases relevant to insulin resistance. Previously, we have identified PTPN9 and DUSP9 as Chebulinic acid potential antidiabetic targets and a multi-targeting natural product thereof. In this study, knockdown of PTPN11 Type 2 diabetes increased AMPK phosphorylation in differentiated C2C12 muscle cells by 3.8 fold, indicating that PTPN11 could Glucose-uptake be an antidiabetic target. Screening of a library of 658 natural products against PTPN9, DUSP9, or PTPN11 PTPN9 identified chebulinic acid (CA) as a strong allosteric inhibitor with a slow cooperative binding toPTPN9 PTPN11 (IC50 = 34 nM) and PTPN11 (IC50 = 37 nM), suggesting that it would be a potential antidiabetic candidate. Furthermore, CA stimulated glucose uptake and resulted in increased AMP-activated protein kinase (AMPK) phosphorylation. Taken together, we demonstrated that CA increased glucose uptake as a dual inhibitor of PTPN9 and PTPN11 through activation of the AMPK signaling pathway. These results strongly suggest that CA could be used as a potential therapeutic candidate for the treatment of type 2 diabetes. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Misplaced Lncrna Causes Brachydactyly in Humans
A misplaced lncRNA causes brachydactyly in humans Philipp G. Maass, … , Friedrich C. Luft, Sylvia Bähring J Clin Invest. 2012;122(11):3990-4002. https://doi.org/10.1172/JCI65508. Research Article Translocations are chromosomal rearrangements that are frequently associated with a variety of disease states and developmental disorders. We identified 2 families with brachydactyly type E (BDE) resulting from different translocations affecting chromosome 12p. Both translocations caused downregulation of the parathyroid hormone-like hormone (PTHLH) gene by disrupting the cis-regulatory landscape. Using chromosome conformation capturing, we identified a regulator on chromosome 12q that interacts in cis with PTHLH over a 24.4-megabase distance and in trans with the sex- determining region Y-box 9 (SOX9) gene on chromosome 17q. The element also harbored a long noncoding RNA (lncRNA). Silencing of the lncRNA, PTHLH, or SOX9 revealed a feedback mechanism involving an expression-dependent network in humans. In the BDE patients, the human lncRNA was upregulated by the disrupted chromosomal association. Moreover, the lncRNA occupancy at the PTHLH locus was reduced. Our results document what we believe to be a novel in cis– and in trans–acting DNA and lncRNA regulatory feedback element that is reciprocally regulated by coding genes. Furthermore, our findings provide a systematic and combinatorial view of how enhancers encoding lncRNAs may affect gene expression in normal development. Find the latest version: https://jci.me/65508/pdf Research article Related Commentary, page 3837 A misplaced lncRNA causes brachydactyly in humans Philipp G. Maass,1,2 Andreas Rump,3 Herbert Schulz,2 Sigmar Stricker,4 Lisanne Schulze,1,2 Konrad Platzer,3 Atakan Aydin,1,2 Sigrid Tinschert,3 Mary B. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Figure S1. Representative Report Generated by the Ion Torrent System Server for Each of the KCC71 Panel Analysis and Pcafusion Analysis
Figure S1. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. A Figure S1. Continued. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. B Figure S2. Comparative analysis of the variant frequency found by the KCC71 panel and calculated from publicly available cBioPortal datasets. For each of the 71 genes in the KCC71 panel, the frequency of variants was calculated as the variant number found in the examined cases. Datasets marked with different colors and sample numbers of prostate cancer are presented in the upper right. *Significantly high in the present study. Figure S3. Seven subnetworks extracted from each of seven public prostate cancer gene networks in TCNG (Table SVI). Blue dots represent genes that include initial seed genes (parent nodes), and parent‑child and child‑grandchild genes in the network. Graphical representation of node‑to‑node associations and subnetwork structures that differed among and were unique to each of the seven subnetworks. TCNG, The Cancer Network Galaxy. Figure S4. REVIGO tree map showing the predicted biological processes of prostate cancer in the Japanese. Each rectangle represents a biological function in terms of a Gene Ontology (GO) term, with the size adjusted to represent the P‑value of the GO term in the underlying GO term database. -

Supp Table 1.Pdf
Upregulated genes in Hdac8 null cranial neural crest cells fold change Gene Symbol Gene Title 134.39 Stmn4 stathmin-like 4 46.05 Lhx1 LIM homeobox protein 1 31.45 Lect2 leukocyte cell-derived chemotaxin 2 31.09 Zfp108 zinc finger protein 108 27.74 0710007G10Rik RIKEN cDNA 0710007G10 gene 26.31 1700019O17Rik RIKEN cDNA 1700019O17 gene 25.72 Cyb561 Cytochrome b-561 25.35 Tsc22d1 TSC22 domain family, member 1 25.27 4921513I08Rik RIKEN cDNA 4921513I08 gene 24.58 Ofa oncofetal antigen 24.47 B230112I24Rik RIKEN cDNA B230112I24 gene 23.86 Uty ubiquitously transcribed tetratricopeptide repeat gene, Y chromosome 22.84 D8Ertd268e DNA segment, Chr 8, ERATO Doi 268, expressed 19.78 Dag1 Dystroglycan 1 19.74 Pkn1 protein kinase N1 18.64 Cts8 cathepsin 8 18.23 1500012D20Rik RIKEN cDNA 1500012D20 gene 18.09 Slc43a2 solute carrier family 43, member 2 17.17 Pcm1 Pericentriolar material 1 17.17 Prg2 proteoglycan 2, bone marrow 17.11 LOC671579 hypothetical protein LOC671579 17.11 Slco1a5 solute carrier organic anion transporter family, member 1a5 17.02 Fbxl7 F-box and leucine-rich repeat protein 7 17.02 Kcns2 K+ voltage-gated channel, subfamily S, 2 16.93 AW493845 Expressed sequence AW493845 16.12 1600014K23Rik RIKEN cDNA 1600014K23 gene 15.71 Cst8 cystatin 8 (cystatin-related epididymal spermatogenic) 15.68 4922502D21Rik RIKEN cDNA 4922502D21 gene 15.32 2810011L19Rik RIKEN cDNA 2810011L19 gene 15.08 Btbd9 BTB (POZ) domain containing 9 14.77 Hoxa11os homeo box A11, opposite strand transcript 14.74 Obp1a odorant binding protein Ia 14.72 ORF28 open reading -
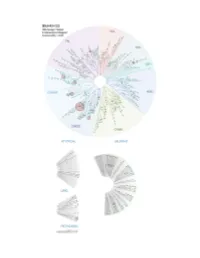
Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) BSJ-03-123 AAK1 AAK1 94 1000 BSJ-03-123 ABL1(E255K)-phosphorylated ABL1 79 1000 BSJ-03-123 ABL1(F317I)-nonphosphorylated ABL1 89 1000 BSJ-03-123 ABL1(F317I)-phosphorylated ABL1 98 1000 BSJ-03-123 ABL1(F317L)-nonphosphorylated ABL1 86 1000 BSJ-03-123 ABL1(F317L)-phosphorylated ABL1 89 1000 BSJ-03-123 ABL1(H396P)-nonphosphorylated ABL1 76 1000 BSJ-03-123 ABL1(H396P)-phosphorylated ABL1 90 1000 BSJ-03-123 ABL1(M351T)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1(Q252H)-nonphosphorylated ABL1 56 1000 BSJ-03-123 ABL1(Q252H)-phosphorylated ABL1 97 1000 BSJ-03-123 ABL1(T315I)-nonphosphorylated ABL1 100 1000 BSJ-03-123 ABL1(T315I)-phosphorylated ABL1 85 1000 BSJ-03-123 ABL1(Y253F)-phosphorylated ABL1 100 1000 BSJ-03-123 ABL1-nonphosphorylated ABL1 60 1000 BSJ-03-123 ABL1-phosphorylated ABL1 79 1000 BSJ-03-123 ABL2 ABL2 89 1000 BSJ-03-123 ACVR1 ACVR1 100 1000 BSJ-03-123 ACVR1B ACVR1B 95 1000 BSJ-03-123 ACVR2A ACVR2A 100 1000 BSJ-03-123 ACVR2B ACVR2B 96 1000 BSJ-03-123 ACVRL1 ACVRL1 84 1000 BSJ-03-123 ADCK3 CABC1 90 1000 BSJ-03-123 ADCK4 ADCK4 91 1000 BSJ-03-123 AKT1 AKT1 100 1000 BSJ-03-123 AKT2 AKT2 98 1000 BSJ-03-123 AKT3 AKT3 100 1000 BSJ-03-123 ALK ALK 100 1000 BSJ-03-123 ALK(C1156Y) ALK 78 1000 BSJ-03-123 ALK(L1196M) ALK 100 1000 BSJ-03-123 AMPK-alpha1 PRKAA1 93 1000 BSJ-03-123 AMPK-alpha2 PRKAA2 100 1000 BSJ-03-123 ANKK1 ANKK1 89 1000 BSJ-03-123 ARK5 NUAK1 98 1000 BSJ-03-123 ASK1 MAP3K5 100 1000 BSJ-03-123 ASK2 MAP3K6 92 1000 BSJ-03-123 AURKA -

ETV4 and AP1 Transcription Factors Form Multivalent Interactions with Three Sites on the MED25 Activator-Interacting Domain
Featured Arcle KDC YJMBI-65461; No. of pages: 21; 4C: ETV4 and AP1 Transcription Factors Form Multivalent Interactions with three Sites on the MED25 Activator-Interacting Domain Simon L. Currie 1,2, Jedediah J. Doane 1,2, Kathryn S. Evans 1,2, Niraja Bhachech 1,2, Bethany J. Madison 1,2, Desmond K.W. Lau 3, Lawrence P. McIntosh 3, Jack J. Skalicky 4, Kathleen A. Clark 1,2 and Barbara J. Graves 1,2,5 1 - Department of Oncological Sciences, University of Utah School of Medicine, Salt Lake City, UT, 84112-5500, USA 2 - Huntsman Cancer Institute, University of Utah, Salt Lake City, UT, 84112-5500, USA 3 - Departments of Biochemistry and Molecular Biology, Department of Chemistry, and Michael Smith Laboratories, University of British Columbia, Vancouver, BC, V6T 1Z3, Canada 4 - Department of Biochemistry, University of Utah School of Medicine, Salt Lake City, UT, 84112-5650, USA 5 - Howard Hughes Medical Institute, Chevy Chase, MD, 20815-6789, USA Correspondence to Barbara J. Graves: Department of Oncological Sciences, University of Utah School of Medicine, Salt Lake City, UT, 84112-5500, USA. [email protected]. http://dx.doi.org/10.1016/j.jmb.2017.06.024 Edited by Prof. M.F. Summers Simon L. Currie, Jedediah J. Doane, Kathryn S. Evans and Barbara J. Graves Abstract The recruitment of transcriptional cofactors by sequence-specific transcription factors challenges the basis of high affinity and selective interactions. Extending previous studies that the N-terminal activation domain (AD) of ETV5 interacts with Mediator subunit 25 (MED25), we establish that similar, aromatic-rich motifs located both in the AD and in the DNA-binding domain (DBD) of the related ETS factor ETV4 interact with MED25. -

IFI35 Is Involved in the Regulation of the Radiosensitivity of Colorectal Cancer Cells
IFI35 is involved in the regulation of the radiosensitivity of colorectal cancer cells Yan Hu The First Peoples of Hospital of Taicang Bing Wang The First Peoples of Hospital of Taicang Ke Yi The First Peoples of Hospital of Taicang Qingjun Lei First Peoples Hospital of Neijiang Guanghui Wang Soochow University Xiaohui Xu ( [email protected] ) The First People's Hospital of Taicang, Taicang Aliated Hospital of Soochow University https://orcid.org/0000-0002-5724-0799 Primary research Keywords: colorectal cancer, IRF1, IFI35, radiosensitivity, luciferase reporter assay Posted Date: May 3rd, 2021 DOI: https://doi.org/10.21203/rs.3.rs-459951/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/20 Abstract Background: Interferon regulatory factor-1 (IRF1) affects the proliferation of colorectal cancer (CRC). Recombinant interferon inducible protein 35 (IFI35) participates in immune regulation and cell proliferation. The aim of the study was to examine whether IRF1 affects the radiation sensitivity of CRC by regulating IFI35. Methods: CCL244 and SW480 cells were divided into ve groups: blank control, IFI35 upregulation, IFI35 upregulation control, IFI35 downregulation, and IFI35 downregulation control. All groups were treated with X-rays (6 Gy). IFI35 activation by IRF1 was detected by luciferase reporter assay. The GEPIA database was used to examine IRF1 and IFI35 in CRC. The cells were characterized using CCK-8, EdU, cell cycle, clone formation, ow cytometry, reactive oxygen species (ROS), and mitochondrial membrane potential. Nude mouse animal models were used to detect the effect of IFI35 on CRC. -

The Regulatory Roles of Phosphatases in Cancer
Oncogene (2014) 33, 939–953 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW The regulatory roles of phosphatases in cancer J Stebbing1, LC Lit1, H Zhang, RS Darrington, O Melaiu, B Rudraraju and G Giamas The relevance of potentially reversible post-translational modifications required for controlling cellular processes in cancer is one of the most thriving arenas of cellular and molecular biology. Any alteration in the balanced equilibrium between kinases and phosphatases may result in development and progression of various diseases, including different types of cancer, though phosphatases are relatively under-studied. Loss of phosphatases such as PTEN (phosphatase and tensin homologue deleted on chromosome 10), a known tumour suppressor, across tumour types lends credence to the development of phosphatidylinositol 3--kinase inhibitors alongside the use of phosphatase expression as a biomarker, though phase 3 trial data are lacking. In this review, we give an updated report on phosphatase dysregulation linked to organ-specific malignancies. Oncogene (2014) 33, 939–953; doi:10.1038/onc.2013.80; published online 18 March 2013 Keywords: cancer; phosphatases; solid tumours GASTROINTESTINAL MALIGNANCIES abs in sera were significantly associated with poor survival in Oesophageal cancer advanced ESCC, suggesting that they may have a clinical utility in Loss of PTEN (phosphatase and tensin homologue deleted on ESCC screening and diagnosis.5 chromosome 10) expression in oesophageal cancer is frequent, Cao et al.6 investigated the role of protein tyrosine phosphatase, among other gene alterations characterizing this disease. Zhou non-receptor type 12 (PTPN12) in ESCC and showed that PTPN12 et al.1 found that overexpression of PTEN suppresses growth and protein expression is higher in normal para-cancerous tissues than induces apoptosis in oesophageal cancer cell lines, through in 20 ESCC tissues.