Reconstructing the Tree of Life
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Taxon Ordering in Phylogenetic Trees by Means of Evolutionary Algorithms Francesco Cerutti1,2, Luigi Bertolotti1,2, Tony L Goldberg3 and Mario Giacobini1,2*
Cerutti et al. BioData Mining 2011, 4:20 http://www.biodatamining.org/content/4/1/20 BioData Mining RESEARCH Open Access Taxon ordering in phylogenetic trees by means of evolutionary algorithms Francesco Cerutti1,2, Luigi Bertolotti1,2, Tony L Goldberg3 and Mario Giacobini1,2* * Correspondence: mario. Abstract [email protected] 1 Department of Animal Production, Background: In in a typical “left-to-right” phylogenetic tree, the vertical order of taxa Epidemiology and Ecology, Faculty of Veterinary Medicine, University is meaningless, as only the branch path between them reflects their degree of of Torino, Via Leonardo da Vinci similarity. To make unresolved trees more informative, here we propose an 44, 10095, Grugliasco (TO), Italy innovative Evolutionary Algorithm (EA) method to search the best graphical Full list of author information is available at the end of the article representation of unresolved trees, in order to give a biological meaning to the vertical order of taxa. Methods: Starting from a West Nile virus phylogenetic tree, in a (1 + 1)-EA we evolved it by randomly rotating the internal nodes and selecting the tree with better fitness every generation. The fitness is a sum of genetic distances between the considered taxon and the r (radius) next taxa. After having set the radius to the best performance, we evolved the trees with (l + μ)-EAs to study the influence of population on the algorithm. Results: The (1 + 1)-EA consistently outperformed a random search, and better results were obtained setting the radius to 8. The (l + μ)-EAs performed as well as the (1 + 1), except the larger population (1000 + 1000). -

Transformations of Lamarckism Vienna Series in Theoretical Biology Gerd B
Transformations of Lamarckism Vienna Series in Theoretical Biology Gerd B. M ü ller, G ü nter P. Wagner, and Werner Callebaut, editors The Evolution of Cognition , edited by Cecilia Heyes and Ludwig Huber, 2000 Origination of Organismal Form: Beyond the Gene in Development and Evolutionary Biology , edited by Gerd B. M ü ller and Stuart A. Newman, 2003 Environment, Development, and Evolution: Toward a Synthesis , edited by Brian K. Hall, Roy D. Pearson, and Gerd B. M ü ller, 2004 Evolution of Communication Systems: A Comparative Approach , edited by D. Kimbrough Oller and Ulrike Griebel, 2004 Modularity: Understanding the Development and Evolution of Natural Complex Systems , edited by Werner Callebaut and Diego Rasskin-Gutman, 2005 Compositional Evolution: The Impact of Sex, Symbiosis, and Modularity on the Gradualist Framework of Evolution , by Richard A. Watson, 2006 Biological Emergences: Evolution by Natural Experiment , by Robert G. B. Reid, 2007 Modeling Biology: Structure, Behaviors, Evolution , edited by Manfred D. Laubichler and Gerd B. M ü ller, 2007 Evolution of Communicative Flexibility: Complexity, Creativity, and Adaptability in Human and Animal Communication , edited by Kimbrough D. Oller and Ulrike Griebel, 2008 Functions in Biological and Artifi cial Worlds: Comparative Philosophical Perspectives , edited by Ulrich Krohs and Peter Kroes, 2009 Cognitive Biology: Evolutionary and Developmental Perspectives on Mind, Brain, and Behavior , edited by Luca Tommasi, Mary A. Peterson, and Lynn Nadel, 2009 Innovation in Cultural Systems: Contributions from Evolutionary Anthropology , edited by Michael J. O ’ Brien and Stephen J. Shennan, 2010 The Major Transitions in Evolution Revisited , edited by Brett Calcott and Kim Sterelny, 2011 Transformations of Lamarckism: From Subtle Fluids to Molecular Biology , edited by Snait B. -

A General Criterion for Translating Phylogenetic Trees Into Linear Sequences
A general criterion for translating phylogenetic trees into linear sequences Proposal (474) to South American Classification Committee In most of the books, papers and check-lists of birds (e.g. Meyer de Schauensee 1970, Stotz et al. 1996; and other taxa e.g. Lewis et al. 2005, Haston et al. 2009, for plants), at least the higher taxa are arranged phylogenetically, with the “oldest” groups (Rheiformes/Tinamiformes) placed first, and the “modern” birds (Passeriformes) at the end. The molecular phylogenetic analysis of Hackett et al. (2008) supports this criterion. For this reason, it would be desirable that in the SACC list not only orders and families, but also genera within families and species within genera, are phylogenetically ordered. In spite of the SACC’s efforts in producing an updated phylogeny-based list, it is evident that there are differences in the way the phylogenetic information has been translated into linear sequences, mainly for node rotation and polytomies. This is probably one of the reasons why Douglas Stotz (Proposal #423) has criticized using sequence to show relationships. He considers that we are creating unstable sequences with little value in terms of understanding of relationships. He added, “We would be much better served by doing what most taxonomic groups do and placing taxa within the hierarchy in alphabetical order, making no pretense that sequence can provide useful information on the branching patterns of trees. This would greatly stabilize sequences and not cost much information about relationships”. However, we encourage creating sequences that reflect phylogeny as much as possible at all taxonomic levels (although we agree with Douglas Stotz that the translation of a phylogenetic tree into a simple linear sequence inevitably involves a loss of information about relationships that is present in the trees on which the sequence is based). -

Name-Bearing Fossil Type Specimens and Taxa Named from National Park Service Areas
Sullivan, R.M. and Lucas, S.G., eds., 2016, Fossil Record 5. New Mexico Museum of Natural History and Science Bulletin 73. 277 NAME-BEARING FOSSIL TYPE SPECIMENS AND TAXA NAMED FROM NATIONAL PARK SERVICE AREAS JUSTIN S. TWEET1, VINCENT L. SANTUCCI2 and H. GREGORY MCDONALD3 1Tweet Paleo-Consulting, 9149 79th Street S., Cottage Grove, MN 55016, -email: [email protected]; 2National Park Service, Geologic Resources Division, 1201 Eye Street, NW, Washington, D.C. 20005, -email: [email protected]; 3Bureau of Land Management, Utah State Office, 440 West 200 South, Suite 500, Salt Lake City, UT 84101: -email: [email protected] Abstract—More than 4850 species, subspecies, and varieties of fossil organisms have been named from specimens found within or potentially within National Park System area boundaries as of the date of this publication. These plants, invertebrates, vertebrates, ichnotaxa, and microfossils represent a diverse collection of organisms in terms of taxonomy, geologic time, and geographic distribution. In terms of the history of American paleontology, the type specimens found within NPS-managed lands, both historically and contemporary, reflect the birth and growth of the science of paleontology in the United States, with many eminent paleontologists among the contributors. Name-bearing type specimens, whether recovered before or after the establishment of a given park, are a notable component of paleontological resources and their documentation is a critical part of the NPS strategy for their management. In this article, name-bearing type specimens of fossil taxa are documented in association with at least 71 NPS administered areas and one former monument, now abolished. -

Evolutionary History of the Butterflyfishes (F: Chaetodontidae
doi:10.1111/j.1420-9101.2009.01904.x Evolutionary history of the butterflyfishes (f: Chaetodontidae) and the rise of coral feeding fishes D. R. BELLWOOD* ,S.KLANTEN*à,P.F.COWMAN* ,M.S.PRATCHETT ,N.KONOW*§ &L.VAN HERWERDEN*à *School of Marine and Tropical Biology, James Cook University, Townsville, Qld, Australia Australian Research Council Centre of Excellence for Coral Reef Studies, James Cook University, Townsville, Qld, Australia àMolecular Evolution and Ecology Laboratory, James Cook University, Townsville, Qld, Australia §Ecology and Evolutionary Biology, Brown University, Providence, RI, USA Keywords: Abstract biogeography; Of the 5000 fish species on coral reefs, corals dominate the diet of just 41 chronogram; species. Most (61%) belong to a single family, the butterflyfishes (Chae- coral reef; todontidae). We examine the evolutionary origins of chaetodontid corallivory innovation; using a new molecular phylogeny incorporating all 11 genera. A 1759-bp molecular phylogeny; sequence of nuclear (S7I1 and ETS2) and mitochondrial (cytochrome b) data trophic novelty. yielded a fully resolved tree with strong support for all major nodes. A chronogram, constructed using Bayesian inference with multiple parametric priors, and recent ecological data reveal that corallivory has arisen at least five times over a period of 12 Ma, from 15.7 to 3 Ma. A move onto coral reefs in the Miocene foreshadowed rapid cladogenesis within Chaetodon and the origins of corallivory, coinciding with a global reorganization of coral reefs and the expansion of fast-growing corals. This historical association underpins the sensitivity of specific butterflyfish clades to global coral decline. butterflyfishes (f. Chaetodontidae); of the remainder Introduction most (eight) are in the Labridae. -

Developmental Sequences of Squamate Reptiles Are Taxon Specific
EVOLUTION & DEVELOPMENT 15:5, 326–343 (2013) DOI: 10.1111/ede.12042 Developmental sequences of squamate reptiles are taxon specific Robin M. Andrews,a,* Matthew C. Brandley,b and Virginia W. Greenea a Department of Biological Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA, 24061, USA b School of Biological Sciences (A08), University of Sydney, Sydney, NSW, 2006, Australia *Author for correspondence (e‐mail: [email protected]) SUMMARY Recent studies in comparative vertebrate plotting the proportions of reconstructed ranks (excluding embryology have focused on two related questions. One unlikely events, PP < 0.05) associated with each event. concerns the existence of a phylotypic period, or indeed any Sequence variability was the lowest towards the middle of period, during development in which sequence variation the phylotypic period and involved three events (allantois among taxa is constrained. The second question concerns contacts chorion, maximum number of pharyngeal slits, and the degree to which developmental characters exhibit a appearance of the apical epidermal ridge [AER]); these events phylogenetic signal. These questions are important because each had only two reconstructed ranks. Squamate clades also they underpin attempts to understand the evolution of differed in the rank order of developmental events. Of the 20 developmental characters and their links to adult morphology. events in our analyses, 12 had strongly supported (PP 0.95) To address these questions, we compared the sequence of sequence ranks that differed at two or more internal nodes of developmental events spanning the so‐called phylotypic the tree. For example, gekkotans are distinguished by the late period of vertebrate development in squamate reptiles (lizards appearance of the allantois bud compared to all other and snakes), from the formation of the primary optic placode to squamates (ranks 7 and 8 vs. -

Definitions in Phylogenetic Taxonomy
Syst. Biol. 48(2):329–351, 1999 Denitions in PhylogeneticTaxonomy:Critique and Rationale PAUL C. SERENO Department of Organismal Biologyand Anatomy, Universityof Chicago, 1027E. 57thStreet, Chicago, Illinois 60637, USA; E-mail: [email protected] Abstract.—Ageneralrationale forthe formulation andplacement of taxonomic denitions in phy- logenetic taxonomyis proposed, andcommonly used terms such as“crown taxon”or “ node-based denition” are more precisely dened. In the formulation of phylogenetic denitions, nested refer- ence taxastabilize taxonomic content. Adenitional conguration termed a node-stem triplet also stabilizes the relationship between the trio of taxaat abranchpoint,in the face of local changein phylogenetic relationships oraddition/ deletion of taxa.Crown-total taxonomiesuse survivorship asa criterion forplacement of node-stem triplets within ataxonomic hierarchy.Diversity,morphol- ogy,andtradition alsoconstitute heuristic criteria forplacement of node-stem triplets. [Content; crown; denition; node;phylogeny; stability; stem; taxonomy.] Doesone type ofphylogenetic denition Mostphylogenetic denitions have been (apomorphy,node,stem) stabilize the taxo- constructedin the systematicliterature since nomiccontent of ataxonmore than another then withoutexplanation or justi cation for in the face oflocal change of relationships? the particulartype ofde nition used. The Isone type of phylogenetic denition more justication given forpreferential use of suitablefor clades with unresolved basalre- node- andstem-based de nitions for crown -
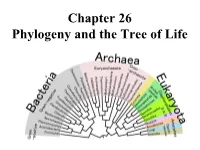
Chapter 26 Phylogeny and the Tree of Life • Biologists Estimate That There Are About 5 to 100 Million Species of Organisms Living on Earth Today
Chapter 26 Phylogeny and the Tree of Life • Biologists estimate that there are about 5 to 100 million species of organisms living on Earth today. • Evidence from morphological, biochemical, and gene sequence data suggests that all organisms on Earth are genetically related, and the genealogical relationships of living things can be represented by a vast evolutionary tree, the Tree of Life. • The Tree of Life then represents the phylogeny of organisms, the history of organismal lineages as they change through time. – In other words, phylogeny is the evolutionary history of a species or group of related species. • Phylogeny assumes that all life arise from a previous ancestors and that all organisms (bacteria, fungi, protist, plants, animals) are connected by the passage of genes along the branches of the phylogenetic tree. • The discipline of systematics classifies organisms and determines their evolutionary relationships. • Systematists use fossil, molecular, and genetic data to infer evolutionary relationships. • Hence, systematists depict evolutionary relationships among organisms as branching phylogenetic trees. • A phylogenetic tree represents a hypothesis about evolutionary relationships. • Taxonomy is the science of organizing, classifying and naming organisms. • Carolus Linnaeus was the scientist who came up with the two-part naming system (binomial system). – The first part of the name is the genus – The second part, called the specific epithet, is the species within the genus. – The first letter of the genus is capitalized, and the entire species name is italicized. • Homo sapiens or H. sapiens • All life are organize into the following taxonomic groups: domain, kingdom, phylum, class, order, family, genus, and species. (Darn Kids Playing Chess On Freeway Gets Squished). -

History of Taxonomy
History of Taxonomy The history of taxonomy dates back to the origin of human language. Western scientific taxonomy started in Greek some hundred years BC and are here divided into prelinnaean and postlinnaean. The most important works are cited and the progress of taxonomy (with the focus on botanical taxonomy) are described up to the era of the Swedish botanist Carl Linnaeus, who founded modern taxonomy. The development after Linnaeus is characterized by a taxonomy that increasingly have come to reflect the paradigm of evolution. The used characters have extended from morphological to molecular. Nomenclatural rules have developed strongly during the 19th and 20th century, and during the last decade traditional nomenclature has been challenged by advocates of the Phylocode. Mariette Manktelow Dept of Systematic Biology Evolutionary Biology Centre Uppsala University Norbyv. 18D SE-752 36 Uppsala E-mail: [email protected] 1. Pre-Linnaean taxonomy 1.1. Earliest taxonomy Taxonomy is as old as the language skill of mankind. It has always been essential to know the names of edible as well as poisonous plants in order to communicate acquired experiences to other members of the family and the tribe. Since my profession is that of a systematic botanist, I will focus my lecture on botanical taxonomy. A taxonomist should be aware of that apart from scientific taxonomy there is and has always been folk taxonomy, which is of great importance in, for example, ethnobiological studies. When we speak about ancient taxonomy we usually mean the history in the Western world, starting with Romans and Greek. However, the earliest traces are not from the West, but from the East. -

Lamarck: the Birth of Biology Author(S): Frans A
Lamarck: The Birth of Biology Author(s): Frans A. Stafleu Reviewed work(s): Source: Taxon, Vol. 20, No. 4 (Aug., 1971), pp. 397-442 Published by: International Association for Plant Taxonomy (IAPT) Stable URL: http://www.jstor.org/stable/1218244 . Accessed: 24/12/2012 16:29 Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at . http://www.jstor.org/page/info/about/policies/terms.jsp . JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range of content in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new forms of scholarship. For more information about JSTOR, please contact [email protected]. International Association for Plant Taxonomy (IAPT) is collaborating with JSTOR to digitize, preserve and extend access to Taxon. http://www.jstor.org This content downloaded on Mon, 24 Dec 2012 16:29:36 PM All use subject to JSTOR Terms and Conditions TAXON 20(4): 397-442. AUGUST 1971 LAMARCK:THE BIRTH OF BIOLOGY Frans A. Stafleu "A long blind patience, such was his genius of the Universe" (Sainte Beuve) Summary A review of the development of Lamarck'sideas on biological systematibswith special reference to the origin and development of his concept of organic evolution. Lamarck's development towards biological systematics is traced through his early botanical and geological writings and related to the gradual change in his scientific outlook from a static and essentialist view of nature towards a dynamic and positivist concept of the life sciences as a special discipline. -

Tetrapod Phylogeny
© J989 Elsevier Science Publishers B. V. (Biomédical Division) The Hierarchy of Life B. Fernholm, K. Bremer and H. Jörnvall, editors 337 CHAPTER 25 Tetrapod phylogeny JACQUES GAUTHIER', DAVID CANNATELLA^, KEVIN DE QUEIROZ^, ARNOLD G. KLUGE* and TIMOTHY ROWE^ ' Deparlmenl qf Herpelology, California Academy of Sciences, San Francisco, CA 94118, U.S.A., ^Museum of Natural Sciences and Department of Biology, Louisiana State University, Baton Rouge, LA 70803, U.S.A., ^Department of ^oology and Museum of Vertebrate ^oology. University of California, Berkeley, CA 94720, U.S.A., 'Museum of ^oology and Department of Biology, University of Michigan, Ann Arbor, MI 48109, U.S.A. and ^Department of Geological Sciences, University of Texas, Austin, TX 78713, U.S.A. Introduction Early sarcopterygians were aquatic, but from the latter part of the Carboniferous on- ward that group has been dominated by terrestrial forms commonly known as the tet- rapods. Fig. 1 illustrates relationships among extant Tetrápoda [1-4J. As the clado- grams in Figs. 2•20 demonstrate, however, extant groups represent only a small part of the taxonomic and morphologic diversity of Tetrápoda. We hope to convey some appreciation for the broad outlines of tetrapod evolution during its 300+ million year history from late Mississippian to Recent times. In doing so, we summarize trees de- rived from the distribution of over 972 characters among 83 terminal taxa of Tetrápo- da. More than 90% of the terminal taxa we discuss are extinct, but all of the subter- minal taxa are represented in the extant biota. This enables us to emphasize the origins of living tetrapod groups while giving due consideration to the diversity and antiquity of the clades of which they are a part. -

Invertebrate-Paleontology-By-Clarkson.Pdf
Invertebrate Palaeontology and Evolution E.N.K. Clarkson Professor of Palaeontology Department of Geology University of Edinburgh Scotland Fourth edition b Blackwell Science Invertebrate Palaeontology and Evolution Invertebrate Palaeontology and Evolution E.N.K. Clarkson Professor of Palaeontology Department of Geology University of Edinburgh Scotland Fourth edition b Blackwell Science © 1979,1986,1993,1998 by E. N. K. Clarkson Published by Blackwell Science Ltd, a Blackwell Publishing company BLACKWELL PUBLlSHING 350 Mam Street, Malden, MA 02148-5020, USA %00 Garsington Road, Oxford OX4 2DQ, UK 550 Swanston Street, Carlton, Victoria 3053, Australia The right of E. :'\I. K. Clarkson to be identified as the Author of this Work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No pad of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, ml'Chanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, WIthout the prior permission of the publisher. First published 1979 by Unwin Hyman Ltd Second edition 1986 Third edition 1993 by Chapman & Hall Fourth edition 1998 by Blackwell Science Ltd 10 2008 Library (If Congress Catalogins-in-Puhlicatioll Unta has bccn applied/or ISBN 978-0-632-05238-7 A catalogue record for this title is available from the British Library. Set by Wyvern 21 Ltd, Bristol Printed and bound in Singapore by C.Os. Printers Pte Ltd The publisher's policy is to use permanent paper from mills that operate a sustainable forestry policy, and which has been manufactured from pulp processed using acid-free and elementary chlorine-free practices.