DFT Calculations on Electronic Density of States and Band Structures of Three Selected Systems Ranel C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![Arxiv:2005.03138V2 [Cond-Mat.Quant-Gas] 23 May 2020 Contents](https://docslib.b-cdn.net/cover/8131/arxiv-2005-03138v2-cond-mat-quant-gas-23-may-2020-contents-58131.webp)
Arxiv:2005.03138V2 [Cond-Mat.Quant-Gas] 23 May 2020 Contents
Condensed Matter Physics in Time Crystals Lingzhen Guo1 and Pengfei Liang2;3 1Max Planck Institute for the Science of Light (MPL), Staudtstrasse 2, 91058 Erlangen, Germany 2Beijing Computational Science Research Center, 100193 Beijing, China 3Abdus Salam ICTP, Strada Costiera 11, I-34151 Trieste, Italy E-mail: [email protected] Abstract. Time crystals are physical systems whose time translation symmetry is spontaneously broken. Although the spontaneous breaking of continuous time- translation symmetry in static systems is proved impossible for the equilibrium state, the discrete time-translation symmetry in periodically driven (Floquet) systems is allowed to be spontaneously broken, resulting in the so-called Floquet or discrete time crystals. While most works so far searching for time crystals focus on the symmetry breaking process and the possible stabilising mechanisms, the many-body physics from the interplay of symmetry-broken states, which we call the condensed matter physics in time crystals, is not fully explored yet. This review aims to summarise the very preliminary results in this new research field with an analogous structure of condensed matter theory in solids. The whole theory is built on a hidden symmetry in time crystals, i.e., the phase space lattice symmetry, which allows us to develop the band theory, topology and strongly correlated models in phase space lattice. In the end, we outline the possible topics and directions for the future research. arXiv:2005.03138v2 [cond-mat.quant-gas] 23 May 2020 Contents 1 Brief introduction to time crystals3 1.1 Wilczek's time crystal . .3 1.2 No-go theorem . .3 1.3 Discrete time-translation symmetry breaking . -

Solid State Physics II Level 4 Semester 1 Course Content
Solid State Physics II Level 4 Semester 1 Course Content L1. Introduction to solid state physics - The free electron theory : Free levels in one dimension. L2. Free electron gas in three dimensions. L3. Electrical conductivity – Motion in magnetic field- Wiedemann-Franz law. L4. Nearly free electron model - origin of the energy band. L5. Bloch functions - Kronig Penney model. L6. Dielectrics I : Polarization in dielectrics L7 .Dielectrics II: Types of polarization - dielectric constant L8. Assessment L9. Experimental determination of dielectric constant L10. Ferroelectrics (1) : Ferroelectric crystals L11. Ferroelectrics (2): Piezoelectricity L12. Piezoelectricity Applications L1 : Solid State Physics Solid state physics is the study of rigid matter, or solids, ,through methods such as quantum mechanics, crystallography, electromagnetism and metallurgy. It is the largest branch of condensed matter physics. Solid-state physics studies how the large-scale properties of solid materials result from their atomic- scale properties. Thus, solid-state physics forms the theoretical basis of materials science. It also has direct applications, for example in the technology of transistors and semiconductors. Crystalline solids & Amorphous solids Solid materials are formed from densely-packed atoms, which interact intensely. These interactions produce : the mechanical (e.g. hardness and elasticity), thermal, electrical, magnetic and optical properties of solids. Depending on the material involved and the conditions in which it was formed , the atoms may be arranged in a regular, geometric pattern (crystalline solids, which include metals and ordinary water ice) , or irregularly (an amorphous solid such as common window glass). Crystalline solids & Amorphous solids The bulk of solid-state physics theory and research is focused on crystals. -

Lecture 3: Fermi-Liquid Theory 1 General Considerations Concerning Condensed Matter
Phys 769 Selected Topics in Condensed Matter Physics Summer 2010 Lecture 3: Fermi-liquid theory Lecturer: Anthony J. Leggett TA: Bill Coish 1 General considerations concerning condensed matter (NB: Ultracold atomic gasses need separate discussion) Assume for simplicity a single atomic species. Then we have a collection of N (typically 1023) nuclei (denoted α,β,...) and (usually) ZN electrons (denoted i,j,...) interacting ∼ via a Hamiltonian Hˆ . To a first approximation, Hˆ is the nonrelativistic limit of the full Dirac Hamiltonian, namely1 ~2 ~2 1 e2 1 Hˆ = 2 2 + NR −2m ∇i − 2M ∇α 2 4πǫ r r α 0 i j Xi X Xij | − | 1 (Ze)2 1 1 Ze2 1 + . (1) 2 4πǫ0 Rα Rβ − 2 4πǫ0 ri Rα Xαβ | − | Xiα | − | For an isolated atom, the relevant energy scale is the Rydberg (R) – Z2R. In addition, there are some relativistic effects which may need to be considered. Most important is the spin-orbit interaction: µ Hˆ = B σ (v V (r )) (2) SO − c2 i · i × ∇ i Xi (µB is the Bohr magneton, vi is the velocity, and V (ri) is the electrostatic potential at 2 3 2 ri as obtained from HˆNR). In an isolated atom this term is o(α R) for H and o(Z α R) for a heavy atom (inner-shell electrons) (produces fine structure). The (electron-electron) magnetic dipole interaction is of the same order as HˆSO. The (electron-nucleus) hyperfine interaction is down relative to Hˆ by a factor µ /µ 10−3, and the nuclear dipole-dipole SO n B ∼ interaction by a factor (µ /µ )2 10−6. -

Phys 446: Solid State Physics / Optical Properties Lattice Vibrations
Solid State Physics Lecture 5 Last week: Phys 446: (Ch. 3) • Phonons Solid State Physics / Optical Properties • Today: Einstein and Debye models for thermal capacity Lattice vibrations: Thermal conductivity Thermal, acoustic, and optical properties HW2 discussion Fall 2007 Lecture 5 Andrei Sirenko, NJIT 1 2 Material to be included in the test •Factors affecting the diffraction amplitude: Oct. 12th 2007 Atomic scattering factor (form factor): f = n(r)ei∆k⋅rl d 3r reflects distribution of electronic cloud. a ∫ r • Crystalline structures. 0 sin()∆k ⋅r In case of spherical distribution f = 4πr 2n(r) dr 7 crystal systems and 14 Bravais lattices a ∫ n 0 ∆k ⋅r • Crystallographic directions dhkl = 2 2 2 1 2 ⎛ h k l ⎞ 2πi(hu j +kv j +lw j ) and Miller indices ⎜ + + ⎟ •Structure factor F = f e ⎜ a2 b2 c2 ⎟ ∑ aj ⎝ ⎠ j • Definition of reciprocal lattice vectors: •Elastic stiffness and compliance. Strain and stress: definitions and relation between them in a linear regime (Hooke's law): σ ij = ∑Cijklε kl ε ij = ∑ Sijklσ kl • What is Brillouin zone kl kl 2 2 C •Elastic wave equation: ∂ u C ∂ u eff • Bragg formula: 2d·sinθ = mλ ; ∆k = G = eff x sound velocity v = ∂t 2 ρ ∂x2 ρ 3 4 • Lattice vibrations: acoustic and optical branches Summary of the Last Lecture In three-dimensional lattice with s atoms per unit cell there are Elastic properties – crystal is considered as continuous anisotropic 3s phonon branches: 3 acoustic, 3s - 3 optical medium • Phonon - the quantum of lattice vibration. Elastic stiffness and compliance tensors relate the strain and the Energy ħω; momentum ħq stress in a linear region (small displacements, harmonic potential) • Concept of the phonon density of states Hooke's law: σ ij = ∑Cijklε kl ε ij = ∑ Sijklσ kl • Einstein and Debye models for lattice heat capacity. -

Chapter 3 Bose-Einstein Condensation of an Ideal
Chapter 3 Bose-Einstein Condensation of An Ideal Gas An ideal gas consisting of non-interacting Bose particles is a ¯ctitious system since every realistic Bose gas shows some level of particle-particle interaction. Nevertheless, such a mathematical model provides the simplest example for the realization of Bose-Einstein condensation. This simple model, ¯rst studied by A. Einstein [1], correctly describes important basic properties of actual non-ideal (interacting) Bose gas. In particular, such basic concepts as BEC critical temperature Tc (or critical particle density nc), condensate fraction N0=N and the dimensionality issue will be obtained. 3.1 The ideal Bose gas in the canonical and grand canonical ensemble Suppose an ideal gas of non-interacting particles with ¯xed particle number N is trapped in a box with a volume V and at equilibrium temperature T . We assume a particle system somehow establishes an equilibrium temperature in spite of the absence of interaction. Such a system can be characterized by the thermodynamic partition function of canonical ensemble X Z = e¡¯ER ; (3.1) R where R stands for a macroscopic state of the gas and is uniquely speci¯ed by the occupa- tion number ni of each single particle state i: fn0; n1; ¢ ¢ ¢ ¢ ¢ ¢g. ¯ = 1=kBT is a temperature parameter. Then, the total energy of a macroscopic state R is given by only the kinetic energy: X ER = "ini; (3.2) i where "i is the eigen-energy of the single particle state i and the occupation number ni satis¯es the normalization condition X N = ni: (3.3) i 1 The probability -

A Short Review of Phonon Physics Frijia Mortuza
International Journal of Scientific & Engineering Research Volume 11, Issue 10, October-2020 847 ISSN 2229-5518 A Short Review of Phonon Physics Frijia Mortuza Abstract— In this article the phonon physics has been summarized shortly based on different articles. As the field of phonon physics is already far ad- vanced so some salient features are shortly reviewed such as generation of phonon, uses and importance of phonon physics. Index Terms— Collective Excitation, Phonon Physics, Pseudopotential Theory, MD simulation, First principle method. —————————— —————————— 1. INTRODUCTION There is a collective excitation in periodic elastic arrangements of atoms or molecules. Melting transition crystal turns into liq- uid and it loses long range transitional order and liquid appears to be disordered from crystalline state. Collective dynamics dispersion in transition materials is mostly studied with a view to existing collective modes of motions, which include longitu- dinal and transverse modes of vibrational motions of the constituent atoms. The dispersion exhibits the existence of collective motions of atoms. This has led us to undertake the study of dynamics properties of different transitional metals. However, this collective excitation is known as phonon. In this article phonon physics is shortly reviewed. 2. GENERATION AND PROPERTIES OF PHONON Generally, over some mean positions the atoms in the crystal tries to vibrate. Even in a perfect crystal maximum amount of pho- nons are unstable. As they are unstable after some time of period they come to on the object surface and enters into a sensor. It can produce a signal and finally it leaves the target object. In other word, each atom is coupled with the neighboring atoms and makes vibration and as a result phonon can be found [1]. -

High Pressure Band Structure, Density of States, Structural Phase Transition and Metallization in Cds
Chemical and Materials Engineering 5(1): 8-13, 2017 http://www.hrpub.org DOI: 10.13189/cme.2017.050102 High Pressure Band Structure, Density of States, Structural Phase Transition and Metallization in CdS J. Jesse Pius1, A. Lekshmi2, C. Nirmala Louis2,* 1Rohini College of Engineering, Nagercoil, Kanyakumari District, India 2Research Center in Physics, Holy Cross College, India Copyright©2017 by authors, all rights reserved. Authors agree that this article remains permanently open access under the terms of the Creative Commons Attribution License 4.0 International License Abstract The electronic band structure, density of high pressure studies due to the development of different states, metallization and structural phase transition of cubic designs of diamond anvil cell (DAC). With a modern DAC, zinc blende type cadmium sulphide (CdS) is investigated it is possible to reach pressures of 2 Mbar (200 GPa) using the full potential linear muffin-tin orbital (FP-LMTO) routinely and pressures of 5 Mbar (500 GPa) or higher is method. The ground state properties and band gap values achievable [3]. At such pressures, materials are reduced to are compared with the experimental results. The fractions of their original volumes. With this reduction in equilibrium lattice constant, bulk modulus and its pressure inter atomic distances; significant changes in bonding and derivative and the phase transition pressure at which the structure as well as other properties take place. The increase compounds undergo structural phase transition from ZnS to of pressure means the significant decrease in volume, which NaCl are predicted from the total energy calculations. The results in the change of electronic states and crystal structure. -
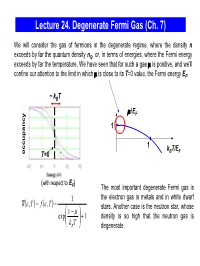
Lecture 24. Degenerate Fermi Gas (Ch
Lecture 24. Degenerate Fermi Gas (Ch. 7) We will consider the gas of fermions in the degenerate regime, where the density n exceeds by far the quantum density nQ, or, in terms of energies, where the Fermi energy exceeds by far the temperature. We have seen that for such a gas μ is positive, and we’ll confine our attention to the limit in which μ is close to its T=0 value, the Fermi energy EF. ~ kBT μ/EF 1 1 kBT/EF occupancy T=0 (with respect to E ) F The most important degenerate Fermi gas is 1 the electron gas in metals and in white dwarf nε()(),, T= f ε T = stars. Another case is the neutron star, whose ε⎛ − μ⎞ exp⎜ ⎟ +1 density is so high that the neutron gas is ⎝kB T⎠ degenerate. Degenerate Fermi Gas in Metals empty states ε We consider the mobile electrons in the conduction EF conduction band which can participate in the charge transport. The band energy is measured from the bottom of the conduction 0 band. When the metal atoms are brought together, valence their outer electrons break away and can move freely band through the solid. In good metals with the concentration ~ 1 electron/ion, the density of electrons in the electron states electron states conduction band n ~ 1 electron per (0.2 nm)3 ~ 1029 in an isolated in metal electrons/m3 . atom The electrons are prevented from escaping from the metal by the net Coulomb attraction to the positive ions; the energy required for an electron to escape (the work function) is typically a few eV. -

Lecture Notes for Quantum Matter
Lecture Notes for Quantum Matter MMathPhys c Professor Steven H. Simon Oxford University July 24, 2019 Contents 1 What we will study 1 1.1 Bose Superfluids (BECs, Superfluid He, Superconductors) . .1 1.2 Theory of Fermi Liquids . .2 1.3 BCS theory of superconductivity . .2 1.4 Special topics . .2 2 Introduction to Superfluids 3 2.1 Some History and Basics of Superfluid Phenomena . .3 2.2 Landau and the Two Fluid Model . .6 2.2.1 More History and a bit of Physics . .6 2.2.2 Landau's Two Fluid Model . .7 2.2.3 More Physical Effects and Their Two Fluid Pictures . .9 2.2.4 Second Sound . 12 2.2.5 Big Questions Remaining . 13 2.3 Curl Free Constraint: Introducing the Superfluid Order Parameter . 14 2.3.1 Vorticity Quantization . 15 2.4 Landau Criterion for Superflow . 17 2.5 Superfluid Density . 20 2.5.1 The Andronikoshvili Experiment . 20 2.5.2 Landau's Calculation of Superfluid Density . 22 3 Charged Superfluid ≈ Superconductor 25 3.1 London Theory . 25 3.1.1 Meissner-Ochsenfeld Effect . 27 3 3.1.2 Quantum Input and Superfluid Order Parameter . 29 3.1.3 Superconducting Vortices . 30 3.1.4 Type I and Type II superconductors . 32 3.1.5 How big is Hc ............................... 33 4 Microscopic Theory of Bosons 37 4.1 Mathematical Preliminaries . 37 4.1.1 Second quantization . 37 4.1.2 Coherent States . 38 4.1.3 Multiple orbitals . 40 4.2 BECs and the Gross-Pitaevskii Equation . 41 4.2.1 Noninteracting BECs as Coherent States . -

Chapter 13 Ideal Fermi
Chapter 13 Ideal Fermi gas The properties of an ideal Fermi gas are strongly determined by the Pauli principle. We shall consider the limit: k T µ,βµ 1, B � � which defines the degenerate Fermi gas. In this limit, the quantum mechanical nature of the system becomes especially important, and the system has little to do with the classical ideal gas. Since this chapter is devoted to fermions, we shall omit in the following the subscript ( ) that we used for the fermionic statistical quantities in the previous chapter. − 13.1 Equation of state Consider a gas ofN non-interacting fermions, e.g., electrons, whose one-particle wave- functionsϕ r(�r) are plane-waves. In this case, a complete set of quantum numbersr is given, for instance, by the three cartesian components of the wave vector �k and thez spin projectionm s of an electron: r (k , k , k , m ). ≡ x y z s Spin-independent Hamiltonians. We will consider only spin independent Hamiltonian operator of the type ˆ 3 H= �k ck† ck + d r V(r)c r†cr , �k � where thefirst and the second terms are respectively the kinetic and th potential energy. The summation over the statesr (whenever it has to be performed) can then be reduced to the summation over states with different wavevectork(p=¯hk): ... (2s + 1) ..., ⇒ r � �k where the summation over the spin quantum numberm s = s, s+1, . , s has been taken into account by the prefactor (2s + 1). − − 159 160 CHAPTER 13. IDEAL FERMI GAS Wavefunctions in a box. We as- sume that the electrons are in a vol- ume defined by a cube with sidesL x, Ly,L z and volumeV=L xLyLz. -

Introduction to Solid State Physics
Introduction to Solid State Physics Sonia Haddad Laboratoire de Physique de la Matière Condensée Faculté des Sciences de Tunis, Université Tunis El Manar S. Haddad, ASP2021-23-07-2021 1 Outline Lecture I: Introduction to Solid State Physics • Brief story… • Solid state physics in daily life • Basics of Solid State Physics Lecture II: Electronic band structure and electronic transport • Electronic band structure: Tight binding approach • Applications to graphene: Dirac electrons Lecture III: Introduction to Topological materials • Introduction to topology in Physics • Quantum Hall effect • Haldane model S. Haddad, ASP2021-23-07-2021 2 It’s an online lecture, but…stay focused… there will be Quizzes and Assignments! S. Haddad, ASP2021-23-07-2021 3 References Introduction to Solid State Physics, Charles Kittel Solid State Physics Neil Ashcroft and N. Mermin Band Theory and Electronic Properties of Solids, John Singleton S. Haddad, ASP2021-23-07-2021 4 Outline Lecture I: Introduction to Solid State Physics • A Brief story… • Solid state physics in daily life • Basics of Solid State Physics Lecture II: Electronic band structure and electronic transport • Tight binding approach • Applications to graphene: Dirac electrons Lecture III: Introduction to Topological materials • Introduction to topology in Physics • Quantum Hall effect • Haldane model S. Haddad, ASP2021-23-07-2021 5 Lecture I: Introduction to solid state Physics What is solid state Physics? Condensed Matter Physics (1960) solids Soft liquids Complex Matter systems Optical lattices, Non crystal Polymers, liquid crystal Biological systems (glasses, crystals, colloids s Economic amorphs) systems Neurosystems… S. Haddad, ASP2021-23-07-2021 6 Lecture I: Introduction to solid state Physics What is condensed Matter Physics? "More is different!" P.W. -

Density of States
Density of states A Material is known to have a high density of states at the Fermi energy. (a) What does this tell you about the electrical, thermal and optical properties of this material? (b) Which of the following quasiparticles would you expect to observe in this material? (phonons, bipolarons, excitons, polaritons, suface plasmons) Why? (a): A high density of states at the fermi energy means that this material is a good electrical conductor. The specific heat can be calculated via the internal energy. Z 1 Z 1 E · D(E) u(E; T ) = E · D(E) · f(E)dE = dE (1) −∞ −∞ 1 + exp E−µ kB ·T du We know that cv = dT . So we get the following expression: E−µ 1 Z E · D(E) · (E − µ) · exp k T c = B dE (2) v 2 −∞ 2 E−µ kBT 1 + exp kbT Hence we deal with a metal, we will have a good thermal conductor because of the phonon and electron contribution. Light will get reflected out below the plasma frequency !P . (b): Due to fact that phonons describe lattice vibrations, it is possible to observe them in this material. They can be measured with an EELS experiment or with Raman Spectroscopy. A polaron describes a local polarisation of a crystal due to moving electrons. They are observable in materials with a low electron density and describe a charge-phonon coupling. There are two different kinds of polarons, the Fr¨ohlich Polaron and the Holstein Polaron. The Fr¨ohlich polarons describe large polarons, that means the distortion is much larger than the lattice constant of the material so that a lot of atoms are involved.