Light Noble Gas Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Primordial Hydrogen-Helium Degassing, an Overlooked Major Energy Source for Internal Terrestrial Processes
HAIT Journal of Science and Engineering B, Volume 2, Issues 1-2, pp. 125-167 Copyright C 2005 Holon Academic Institute of Technology ° Primordial hydrogen-helium degassing, an overlooked major energy source for internal terrestrial processes 1, 2 Arie Gilat ∗ and Alexander Vol 1Geological Survey of Israel (ret.), 8 Yehoash St., Jerusalem 93152, Israel 2Enlightment Ltd. 33 David Pinsky St., Haifa 34454, Israel ∗Corresponding author: [email protected] Received 20 July 2004, revised 3 February 2005, accepted 16 February 2005 Abstract The currently accepted theories concerning terrestrial processes are lacking in accounting for a source of internal energy which: (a) are quickly focused, e.g. earthquakes and volcanic eruptions; (b) are of very high density; (c) provide very high velocities of energy re- lease; (d) have very high density of the energy transport and relatively small losses during transportation over long distances; (e) are quasi- constantly released and practically limitless. This energy release is always accompanied by H- and He-degassing. Solid solutions of H and He, and compounds of He with H, O, Si and metals were discovered in laboratory experiments of ultra-high PT-conditions; He-S, He-Cl, He- C, He-N structures can be deduced from their atomic structure and compositions of natural He-reach gases. Ultra-high PT-conditions ex- ist in the Earth’s interior; hence it seems most likely that some “exotic” compounds are present in the Earth’s core and mantle. During Earth’s accretion, primordial hydrogen and helium were trapped and stored in the planet interior as H- and He- solutions and compounds, stable only under ultrahigh PT-conditions that were dis- covered in recent experiments. -

An Alternate Graphical Representation of Periodic Table of Chemical Elements Mohd Abubakr1, Microsoft India (R&D) Pvt
An Alternate Graphical Representation of Periodic table of Chemical Elements Mohd Abubakr1, Microsoft India (R&D) Pvt. Ltd, Hyderabad, India. [email protected] Abstract Periodic table of chemical elements symbolizes an elegant graphical representation of symmetry at atomic level and provides an overview on arrangement of electrons. It started merely as tabular representation of chemical elements, later got strengthened with quantum mechanical description of atomic structure and recent studies have revealed that periodic table can be formulated using SO(4,2) SU(2) group. IUPAC, the governing body in Chemistry, doesn‟t approve any periodic table as a standard periodic table. The only specific recommendation provided by IUPAC is that the periodic table should follow the 1 to 18 group numbering. In this technical paper, we describe a new graphical representation of periodic table, referred as „Circular form of Periodic table‟. The advantages of circular form of periodic table over other representations are discussed along with a brief discussion on history of periodic tables. 1. Introduction The profoundness of inherent symmetry in nature can be seen at different depths of atomic scales. Periodic table symbolizes one such elegant symmetry existing within the atomic structure of chemical elements. This so called „symmetry‟ within the atomic structures has been widely studied from different prospects and over the last hundreds years more than 700 different graphical representations of Periodic tables have emerged [1]. Each graphical representation of chemical elements attempted to portray certain symmetries in form of columns, rows, spirals, dimensions etc. Out of all the graphical representations, the rectangular form of periodic table (also referred as Long form of periodic table or Modern periodic table) has gained wide acceptance. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

Midterm Examination #3, December 11, 2015 1. (10 Point
NAME: NITROMETHANE CHEMISTRY 443, Fall, 2015(15F) Section Number: 10 Midterm Examination #3, December 11, 2015 Answer each question in the space provided; use back of page if extra space is needed. Answer questions so the grader can READILY understand your work; only work on the exam sheet will be considered. Write answers, where appropriate, with reasonable numbers of significant figures. You may use only the "Student Handbook," a calculator, and a straight edge. 1. (10 points) Argon is a noble gas. For all practical purposes it can be considered an ideal gas. DO NOT WRITE Calculate the change in molar entropy of argon when it is subjected to a process in which the molar IN THIS SPACE volume is tripled and the temperature is simultaneously changed from 300 K to 400 K. 1,2 _______/25 This is a straightforward application of thermodynamics: 3,4 _______/25 = = + = + 2 2 2 2 5 _______/20 Identifying ∆the derivative� and� doing1 � the� integrals � 1give� � �1 � � �1 3 3 400 6,7 _______/20 = + = 8.3144349 + 8.3144349 2 2 1 2 300 8 _______/10 where the∆ heat capacity � 1� at constant � 1volume� of a monatomic �ideal� gas is� . � � � 3 ============= = 9.13434 + 3.58786 = 12.722202 9 _______/5 ∆ (Extra credit) ============= TOTAL PTS 2. (15 points) Benzene ( • = 96.4 ) and toluene ( • = 28.9 ) form a nearly ideal solution over a wide range. For purposes of this question, you may assume that a solution of the two is ideal. (a) What is the total vapor pressure above a solution containing 5.00 moles of benzene and 3.25 moles of toluene? 5.00 3.25 = • + • = (96.4 ) + (28.9 ) 5.00 + 3.25 5.00 + 3.25 = 58.4 + 11.4 = 69.8 (b) What is mole fraction of benzene in the vapor above this solution? . -

The Oganesson Odyssey Kit Chapman Explores the Voyage to the Discovery of Element 118, the Pioneer Chemist It Is Named After, and False Claims Made Along the Way
in your element The oganesson odyssey Kit Chapman explores the voyage to the discovery of element 118, the pioneer chemist it is named after, and false claims made along the way. aving an element named after you Ninov had been dismissed from Berkeley for is incredibly rare. In fact, to be scientific misconduct in May5, and had filed Hhonoured in this manner during a grievance procedure6. your lifetime has only happened to Today, the discovery of the last element two scientists — Glenn Seaborg and of the periodic table as we know it is Yuri Oganessian. Yet, on meeting Oganessian undisputed, but its structure and properties it seems fitting. A colleague of his once remain a mystery. No chemistry has been told me that when he first arrived in the performed on this radioactive giant: 294Og halls of Oganessian’s programme at the has a half-life of less than a millisecond Joint Institute for Nuclear Research (JINR) before it succumbs to α -decay. in Dubna, Russia, it was unlike anything Theoretical models however suggest it he’d ever experienced. Forget the 2,000 ton may not conform to the periodic trends. As magnets, the beam lines and the brand new a noble gas, you would expect oganesson cyclotron being installed designed to hunt to have closed valence shells, ending with for elements 119 and 120, the difference a filled 7s27p6 configuration. But in 2017, a was Oganessian: “When you come to work US–New Zealand collaboration predicted for Yuri, it’s not like a lab,” he explained. that isn’t the case7. -

Helium Adsorption on Lithium Substrates
JLowTempPhys DOI 10.1007/s10909-007-9516-5 Helium Adsorption on Lithium Substrates E. Van Cleve · P. Taborek · J.E. Rutledge Received: 25 July 2007 / Accepted: 13 September 2007 © Springer Science+Business Media 2007 Abstract We have developed a cryogenic pulsed laser deposition (PLD) system to deposit lithium films onto a quartz crystal microbalance (QCM) at 4 K. Adsorption isotherms of 4He on lithium were measured in the temperature range between 1.42 K and 2.5 K. The isotherms are qualitatively different from isotherms on strong sub- strates such as gold and weak substrates such as cesium. There is no evidence of the formation of solid-like layers of helium, and the helium coverage is approximately linear in the pressure over a wide range. By measuring the low coverage slope of the isotherms, the binding energy of helium to lithium was found to be approxi- mately −13.6 K. For lithium substrates less than approximately 100 layers thick, the chemical potential at which the superfluid transition was observed was surprisingly sensitive to the details of lithium deposition. Keywords Helium films · Pulsed laser deposition · Superfluidity · Alkali metal 1 Introduction When helium is adsorbed onto a strong heterogenous substrate such as gold, the first 2 or 3 statistical layers are solid-like. The nature of these layers is not yet clear, but the layers are amorphous and do not participate significantly in superflow at high coverages. Superfluidity on strong substrates requires a minimum critical coverage to saturate the solid-like layers, and the superfluid phase which forms at higher cover- ages flows over these layers and does not interact directly with the strong, short range This work was supported by NSF grant DMR 0509685. -

Of the Periodic Table
of the Periodic Table teacher notes Give your students a visual introduction to the families of the periodic table! This product includes eight mini- posters, one for each of the element families on the main group of the periodic table: Alkali Metals, Alkaline Earth Metals, Boron/Aluminum Group (Icosagens), Carbon Group (Crystallogens), Nitrogen Group (Pnictogens), Oxygen Group (Chalcogens), Halogens, and Noble Gases. The mini-posters give overview information about the family as well as a visual of where on the periodic table the family is located and a diagram of an atom of that family highlighting the number of valence electrons. Also included is the student packet, which is broken into the eight families and asks for specific information that students will find on the mini-posters. The students are also directed to color each family with a specific color on the blank graphic organizer at the end of their packet and they go to the fantastic interactive table at www.periodictable.com to learn even more about the elements in each family. Furthermore, there is a section for students to conduct their own research on the element of hydrogen, which does not belong to a family. When I use this activity, I print two of each mini-poster in color (pages 8 through 15 of this file), laminate them, and lay them on a big table. I have students work in partners to read about each family, one at a time, and complete that section of the student packet (pages 16 through 21 of this file). When they finish, they bring the mini-poster back to the table for another group to use. -

AZMAN AIR CLASSES DANGEROUS GOODS 'Dangerous Goods' Are
AZMAN AIR CLASSES DANGEROUS GOODS ‘Dangerous goods’ are materials or items with hazardous properties Commonly Transported Explosives which, if not properly controlled, 1. Ammunition/cartridges present a potential hazard to human 2. Fireworks/pyrotechnics health and safety, infrastructure and/ 3. Flares or their means of transport. 4. Blasting caps / detonators 5. Fuse The transportation of dangerous 6. Primers goods is controlled and governed by a 7. Explosive charges (blasting, variety of different regulatory demolition etc) regimes, operating at both the 8. Detonating cord national and international levels. 9. Air bag inflators Prominent regulatory frameworks for 10. Igniters the transportation of dangerous 11. Rockets goods include the United Nations 12. TNT / TNT compositions Recommendations on the Transport 13. RDX / RDX compositions of Dangerous Goods, ICAO’s Technical 14. PETN / PETN compositions Instructions, IATA’s Dangerous Goods Regulations and the IMO’s International Maritime Dangerous Commonly Transported Gases Goods Code. Collectively, these 1. Aerosols regulatory regimes mandate how 2. Compressed air dangerous goods are to be handled, 3. Hydrocarbon gas-powered packaged, labelled and transported. devices 1. Explosives 4. Fire extinguishers 2. Gases 5. Gas cartridges 3. Flammable Liquids 6. Fertilizer ammoniating 4. Flammable Solids solution 5. Oxidizing Substances 7. Insecticide gases 6. Toxic & Infectious Substances 8. Refrigerant gases 7. Radioactive Material 9. Lighters 8. Corrosives 10. Acetylene / Oxyacetylene 9. Miscellaneous Dangerous 11. Carbon dioxide Goods 12. Helium / helium compounds 13. Hydrogen / hydrogen compounds 14. Oxygen / oxygen 21. Organochlorine pesticides compounds 22. Organophosphorus 15. Nitrogen / nitrogen pesticides compounds 23. Copper based pesticides 16. Natural gas 24. Esters 17. Oil gas 25. Ethers 18. -

Three Related Topics on the Periodic Tables of Elements
Three related topics on the periodic tables of elements Yoshiteru Maeno*, Kouichi Hagino, and Takehiko Ishiguro Department of physics, Kyoto University, Kyoto 606-8502, Japan * [email protected] (The Foundations of Chemistry: received 30 May 2020; accepted 31 July 2020) Abstaract: A large variety of periodic tables of the chemical elements have been proposed. It was Mendeleev who proposed a periodic table based on the extensive periodic law and predicted a number of unknown elements at that time. The periodic table currently used worldwide is of a long form pioneered by Werner in 1905. As the first topic, we describe the work of Pfeiffer (1920), who refined Werner’s work and rearranged the rare-earth elements in a separate table below the main table for convenience. Today’s widely used periodic table essentially inherits Pfeiffer’s arrangements. Although long-form tables more precisely represent electron orbitals around a nucleus, they lose some of the features of Mendeleev’s short-form table to express similarities of chemical properties of elements when forming compounds. As the second topic, we compare various three-dimensional helical periodic tables that resolve some of the shortcomings of the long-form periodic tables in this respect. In particular, we explain how the 3D periodic table “Elementouch” (Maeno 2001), which combines the s- and p-blocks into one tube, can recover features of Mendeleev’s periodic law. Finally we introduce a topic on the recently proposed nuclear periodic table based on the proton magic numbers (Hagino and Maeno 2020). Here, the nuclear shell structure leads to a new arrangement of the elements with the proton magic-number nuclei treated like noble-gas atoms. -
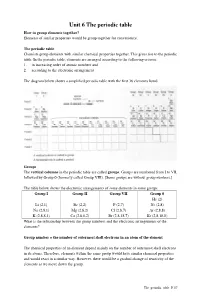
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -

The Noble Gases
INTERCHAPTER K The Noble Gases When an electric discharge is passed through a noble gas, light is emitted as electronically excited noble-gas atoms decay to lower energy levels. The tubes contain helium, neon, argon, krypton, and xenon. University Science Books, ©2011. All rights reserved. www.uscibooks.com Title General Chemistry - 4th ed Author McQuarrie/Gallogy Artist George Kelvin Figure # fig. K2 (965) Date 09/02/09 Check if revision Approved K. THE NOBLE GASES K1 2 0 Nitrogen and He Air P Mg(ClO ) NaOH 4 4 2 noble gases 4.002602 1s2 O removal H O removal CO removal 10 0 2 2 2 Ne Figure K.1 A schematic illustration of the removal of O2(g), H2O(g), and CO2(g) from air. First the oxygen is removed by allowing the air to pass over phosphorus, P (s) + 5 O (g) → P O (s). 20.1797 4 2 4 10 2s22p6 The residual air is passed through anhydrous magnesium perchlorate to remove the water vapor, Mg(ClO ) (s) + 6 H O(g) → Mg(ClO ) ∙6 H O(s), and then through sodium hydroxide to remove 18 0 4 2 2 4 2 2 the carbon dioxide, NaOH(s) + CO2(g) → NaHCO3(s). The gas that remains is primarily nitrogen Ar with about 1% noble gases. 39.948 3s23p6 36 0 The Group 18 elements—helium, K-1. The Noble Gases Were Kr neon, argon, krypton, xenon, and Not Discovered until 1893 83.798 radon—are called the noble gases 2 6 4s 4p and are noteworthy for their rela- In 1893, the English physicist Lord Rayleigh noticed 54 0 tive lack of chemical reactivity.