Guidance Document Validation of Chlorine and Oxygen Isotope Ratio Analysis to Differentiate Perchlorate Sources and to Document Perchlorate Biodegradation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Chapter 8: Ionic Compounds
210-239_Ch08-866418 5/8/06 5:22 PM Page 210 CHAPTER 8 Ionic Compounds Chemistry 1.d, 1.g, 1.i, 2.a, 2.c, 2.e, 7.b I&E 1.b, 1.c, 1.d, 1.l What You’ll Learn ▲ You will define a chemical bond. ▲ You will describe how ions form. ▲ You will identify ionic bonding and the character- istics of ionic compounds. ▲ You will name and write formulas for ionic compounds. ▲ You will relate metallic bonds to the characteristics of metals. Why It’s Important The world around you is com- posed mainly of compounds. The properties of each com- pound are based on how the compound is bonded. The salts dissolved in Earth’s oceans and the compounds that make up most of Earth’s crust are held together by ionic bonds. Visit the Chemistry Web site at chemistrymc.com to find links about ionic compounds. The rock surface, the climbers’ equipment, the atmosphere, and even the climbers are composed almost entirely of compounds and mixtures of compounds. 210 Chapter 8 210-239_Ch08-866418 5/8/06 5:22 PM Page 211 DISCOVERY LAB An Unusual Alloy ost metals that you encounter are solids. Can a metal melt at a Mtemperature below the boiling point of water? You will use a metal alloy called Onion’s Fusible Alloy to answer this question. Safety Precautions Use caution around the heat source and the heated beaker and its contents. Procedure 1. Carefully place a small piece of Onion’s Fusible Alloy into a 250-mL beaker. -
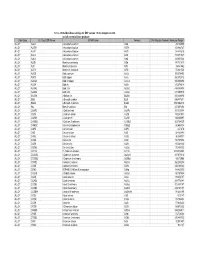
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -
Standard X-Ray Diffraction Powder Patterns
E^l Admin. NBS MONOGRAPH 25—SECTION 5 Refecii^M not to be ^ferlrom the library. Standard X-ray Diffraction Powder Patterns ^\ / U.S. DEPARTMENT OF COMMERCE S NATIONAL BUREAU OF STANDARDS THE NATIONAL BUREAU OF STANDARDS The National Bureau of Standards^ provides measurement and technical information services essential to the efficiency and effectiveness of the work of the Nation's scientists and engineers. The Bureau serves also as a focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. To accomplish this mission, the Bureau is organized into three institutes covering broad program areas of research and services: THE INSTITUTE FOR BASIC STANDARDS . provides the central basis within the United States for a complete and consistent system of physical measurements, coordinates that system with the measurement systems of other nations, and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry, and commerce. This Institute comprises a series of divisions, each serving a classical subject matter area: —Applied Mathematics—Electricity—Metrology—Mechanics—Heat—Atomic Physics—Physical Chemistry—Radiation Physics— -Laboratory Astrophysics^—Radio Standards Laboratory,^ which includes Radio Standards Physics and Radio Standards Engineering—Office of Standard Refer- ence Data. THE INSTITUTE FOR MATERIALS RESEARCH . conducts materials research and provides associated materials services including mainly reference materials and data on the properties of ma- terials. Beyond its direct interest to the Nation's scientists and engineers, this Institute yields services which are essential to the advancement of technology in industry and commerce. -

Physical Properties of Chemicals in PAC Revision 27 Listing
LLNL-TR-625492 Physical Properties of Chemicals in PAC Revision 27 Listing M. A. Johnson March 8, 2013 Disclaimer This document was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor Lawrence Livermore National Security, LLC, nor any of their employees makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States government or Lawrence Livermore National Security, LLC. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or Lawrence Livermore National Security, LLC, and shall not be used for advertising or product endorsement purposes. This work performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Physical Properties of Chemicals in PAC Revision 27 Listing 1 Purpose The purpose of this chemical physical property listing is to provide data required to apply the DOE SCAPA Protective Action Criteria (PAC) values to calculation of the LLNL Quantity (Q) Value thresholds for facility chemical hazard classification. This chemical physical property listing based on the DOE SCAPA Protective Action Criteria (PAC) Revision 27 listing Identifies: 1. Physical state at 25°C (i.e. -

United States Patent (19) 11) 4,114,064 Ernsthausen 45) Sep
United States Patent (19) 11) 4,114,064 Ernsthausen 45) Sep. 12, 1978 54 MULTIPLEGASEOUS DISCHARGE Walter H. Kohl, Reinhold Publishing Corp., New DISPLAYAMEMORY PANEL HAVING York, N.Y. 1962, pp. 24, 53, 54, 72-75. IMPROVED VOLTAGE CHARACTERISTICS Primary Examiner-Palmer C. Demeo 75 Inventor: Roger E. Ernsthausen, Luckey, Ohio Attorney, Agent, or Firm-Donald K. Wedding; Myron 73) Assignee: Owens-Illinois, Inc., Toledo, Ohio E. Click; David H. Wilson (21) Appl. No.: 293,817 (57) ABSTRACT 22 Filed: Oct. 2, 1972 There is disclosed a multiple gaseous discharge dis play/memory panel having an electrical memory and Related U.S. Application Data capable of producing a visual display, the panel being (63) Continuation-in-part of Ser. No. 60,376, Aug. 3, 1970, characterized by an ionizable gaseous medium in a gas abandoned. chamber formed by a pair of opposed dielectric material 51 Int. C.’........................ H01J 61/30; H01J 61/35 charge storage members which are respectively backed 52 U.S. C. .................................... 313/188; 313/201; by an array of electrode (conductor) members such that 313/220; 313/221 each array of electrode members is insulated from the 58) Field of Search ................ 313/188,221, 220, 201 gaseous medium by a dielectric member, the electrode members behind each dielectric material member being 56) References Cited oriented with respect to the electrode members behind U.S. PATENT DOCUMENTS the opposing dielectric material member so as to define 2,020,727 11/1935 Gaidies et al. ....................... 313/213 a plurality of discrete discharge volumes, each dis 3,189,781 6/1965 Lempert ..........