5'-Proximal Cis-Acting RNA Signals for Coronavirus Genome Replication
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

On the Coronaviruses and Their Associations with the Aquatic Environment and Wastewater
water Review On the Coronaviruses and Their Associations with the Aquatic Environment and Wastewater Adrian Wartecki 1 and Piotr Rzymski 2,* 1 Faculty of Medicine, Poznan University of Medical Sciences, 60-812 Pozna´n,Poland; [email protected] 2 Department of Environmental Medicine, Poznan University of Medical Sciences, 60-806 Pozna´n,Poland * Correspondence: [email protected] Received: 24 April 2020; Accepted: 2 June 2020; Published: 4 June 2020 Abstract: The outbreak of Coronavirus Disease 2019 (COVID-19), a severe respiratory disease caused by betacoronavirus SARS-CoV-2, in 2019 that further developed into a pandemic has received an unprecedented response from the scientific community and sparked a general research interest into the biology and ecology of Coronaviridae, a family of positive-sense single-stranded RNA viruses. Aquatic environments, lakes, rivers and ponds, are important habitats for bats and birds, which are hosts for various coronavirus species and strains and which shed viral particles in their feces. It is therefore of high interest to fully explore the role that aquatic environments may play in coronavirus spread, including cross-species transmissions. Besides the respiratory tract, coronaviruses pathogenic to humans can also infect the digestive system and be subsequently defecated. Considering this, it is pivotal to understand whether wastewater can play a role in their dissemination, particularly in areas with poor sanitation. This review provides an overview of the taxonomy, molecular biology, natural reservoirs and pathogenicity of coronaviruses; outlines their potential to survive in aquatic environments and wastewater; and demonstrates their association with aquatic biota, mainly waterfowl. It also calls for further, interdisciplinary research in the field of aquatic virology to explore the potential hotspots of coronaviruses in the aquatic environment and the routes through which they may enter it. -

The COVID-19 Pandemic: a Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control
Journal of Clinical Medicine Review The COVID-19 Pandemic: A Comprehensive Review of Taxonomy, Genetics, Epidemiology, Diagnosis, Treatment, and Control Yosra A. Helmy 1,2,* , Mohamed Fawzy 3,*, Ahmed Elaswad 4, Ahmed Sobieh 5, Scott P. Kenney 1 and Awad A. Shehata 6,7 1 Department of Veterinary Preventive Medicine, Ohio Agricultural Research and Development Center, The Ohio State University, Wooster, OH 44691, USA; [email protected] 2 Department of Animal Hygiene, Zoonoses and Animal Ethology, Faculty of Veterinary Medicine, Suez Canal University, Ismailia 41522, Egypt 3 Department of Virology, Faculty of Veterinary Medicine, Suez Canal University, Ismailia 41522, Egypt 4 Department of Animal Wealth Development, Faculty of Veterinary Medicine, Suez Canal University, Ismailia 41522, Egypt; [email protected] 5 Department of Radiology, University of Massachusetts Medical School, Worcester, MA 01655, USA; [email protected] 6 Avian and Rabbit Diseases Department, Faculty of Veterinary Medicine, Sadat City University, Sadat 32897, Egypt; [email protected] 7 Research and Development Section, PerNaturam GmbH, 56290 Gödenroth, Germany * Correspondence: [email protected] (Y.A.H.); [email protected] (M.F.) Received: 18 March 2020; Accepted: 21 April 2020; Published: 24 April 2020 Abstract: A pneumonia outbreak with unknown etiology was reported in Wuhan, Hubei province, China, in December 2019, associated with the Huanan Seafood Wholesale Market. The causative agent of the outbreak was identified by the WHO as the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), producing the disease named coronavirus disease-2019 (COVID-19). The virus is closely related (96.3%) to bat coronavirus RaTG13, based on phylogenetic analysis. -

Identification of New Respiratory Viruses in the New Millennium
Viruses 2015, 7, 996-1019; doi:10.3390/v7030996 OPEN ACCESS viruses ISSN 1999-4915 www.mdpi.com/journal/viruses Review Identification of New Respiratory Viruses in the New Millennium Michael Berry 1,2, Junaid Gamieldien 1 and Burtram C. Fielding 2,* 1 South African National Bioinformatics Institute, University of the Western Cape, Western Cape 7535, South Africa; E-Mails: [email protected] (M.B.); [email protected] (J.G.) 2 Molecular Biology and Virology Laboratory, Department of Medical BioSciences, Faculty of Natural Sciences, University of the Western Cape, Western Cape 7535, South Africa * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +27-21-959-3620; Fax: +27-21-959-3125. Academic Editor: Eric O. Freed Received: 11 December 2014 / Accepted: 26 February 2015 / Published: 6 March 2015 Abstract: The rapid advancement of molecular tools in the past 15 years has allowed for the retrospective discovery of several new respiratory viruses as well as the characterization of novel emergent strains. The inability to characterize the etiological origins of respiratory conditions, particularly in children, led several researchers to pursue the discovery of the underlying etiology of disease. In 2001, this led to the discovery of human metapneumovirus (hMPV) and soon following that the outbreak of Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) promoted an increased interest in coronavirology and the latter discovery of human coronavirus (HCoV) NL63 and HCoV-HKU1. Human bocavirus, with its four separate lineages, discovered in 2005, has been linked to acute respiratory tract infections and gastrointestinal complications. Middle East Respiratory Syndrome coronavirus (MERS-CoV) represents the most recent outbreak of a completely novel respiratory virus, which occurred in Saudi Arabia in 2012 and presents a significant threat to human health. -

Drawing Comparisons Between SARS-Cov-2 and the Animal Coronaviruses
microorganisms Review Drawing Comparisons between SARS-CoV-2 and the Animal Coronaviruses Souvik Ghosh 1,* and Yashpal S. Malik 2 1 Department of Biomedical Sciences, Ross University School of Veterinary Medicine, Basseterre 334, Saint Kitts and Nevis 2 College of Animal Biotechnology, Guru Angad Dev Veterinary and Animal Science University, Ludhiana 141004, India; [email protected] * Correspondence: [email protected] or [email protected]; Tel.: +1-869-4654161 (ext. 401-1202) Received: 23 September 2020; Accepted: 19 November 2020; Published: 23 November 2020 Abstract: The COVID-19 pandemic, caused by a novel zoonotic coronavirus (CoV), SARS-CoV-2, has infected 46,182 million people, resulting in 1,197,026 deaths (as of 1 November 2020), with devastating and far-reaching impacts on economies and societies worldwide. The complex origin, extended human-to-human transmission, pathogenesis, host immune responses, and various clinical presentations of SARS-CoV-2 have presented serious challenges in understanding and combating the pandemic situation. Human CoVs gained attention only after the SARS-CoV outbreak of 2002–2003. On the other hand, animal CoVs have been studied extensively for many decades, providing a plethora of important information on their genetic diversity, transmission, tissue tropism and pathology, host immunity, and therapeutic and prophylactic strategies, some of which have striking resemblance to those seen with SARS-CoV-2. Moreover, the evolution of human CoVs, including SARS-CoV-2, is intermingled with those of animal CoVs. In this comprehensive review, attempts have been made to compare the current knowledge on evolution, transmission, pathogenesis, immunopathology, therapeutics, and prophylaxis of SARS-CoV-2 with those of various animal CoVs. -

Occurrence and Significance of Human Coronaviruses and Human Bocaviruses in Acute Gastroenteritis of Childhood AUT 2153 MINNA PALONIEMI
MINNA PALONIEMI Occurrence and Significance of Human Coronaviruses and Human Bocaviruses in Acute ... Occurrence and Significance of Human Coronaviruses Bocaviruses in PALONIEMI MINNA Acta Universitatis Tamperensis 2153 MINNA PALONIEMI Occurrence and Significance of Human Coronaviruses and Human Bocaviruses in Acute Gastroenteritis of Childhood AUT 2153 AUT MINNA PALONIEMI Occurrence and Significance of Human Coronaviruses and Human Bocaviruses in Acute Gastroenteritis of Childhood ACADEMIC DISSERTATION To be presented, with the permission of the Board of the School of Medicine of the University of Tampere, for public discussion in the Jarmo Visakorpi auditorium of the Arvo building, Lääkärinkatu 1, Tampere, on 15 April 2016, at 12 o’clock. UNIVERSITY OF TAMPERE MINNA PALONIEMI Occurrence and Significance of Human Coronaviruses and Human Bocaviruses in Acute Gastroenteritis of Childhood Acta Universitatis Tamperensis 2153 Tampere University Press Tampere 2016 ACADEMIC DISSERTATION University of Tampere, School of Medicine Vaccine Research Center Finland Supervised by Reviewed by Professor emeritus Timo Vesikari Docent Tapani Hovi University of Tampere University of Helsinki Finland Finland Professor emeritus Olli Ruuskanen University of Turku Finland The originality of this thesis has been checked using the Turnitin OriginalityCheck service in accordance with the quality management system of the University of Tampere. Copyright ©2016 Tampere University Press and the author Cover design by Mikko Reinikka Distributor: [email protected] https://verkkokauppa.juvenes.fi Acta Universitatis Tamperensis 2153 Acta Electronica Universitatis Tamperensis 1652 ISBN 978-952-03-0078-4 (print) ISBN 978-952-03-0079-1 (pdf) ISSN-L 1455-1616 ISSN 1456-954X ISSN 1455-1616 http://tampub.uta.fi Suomen Yliopistopaino Oy – Juvenes Print Tampere 2016 441 729 Painotuote To my family, Abstract Acute gastroenteritis (AGE) remains an important cause of child hospitalization in Finland, although rotavirus vaccination has reduced the number of AGE hospitalizations significantly. -

Betacoronavirus Genomes: How Genomic Information Has Been Used to Deal with Past Outbreaks and the COVID-19 Pandemic
International Journal of Molecular Sciences Review Betacoronavirus Genomes: How Genomic Information Has Been Used to Deal with Past Outbreaks and the COVID-19 Pandemic Alejandro Llanes 1 , Carlos M. Restrepo 1 , Zuleima Caballero 1 , Sreekumari Rajeev 2 , Melissa A. Kennedy 3 and Ricardo Lleonart 1,* 1 Centro de Biología Celular y Molecular de Enfermedades, Instituto de Investigaciones Científicas y Servicios de Alta Tecnología (INDICASAT AIP), Panama City 0801, Panama; [email protected] (A.L.); [email protected] (C.M.R.); [email protected] (Z.C.) 2 College of Veterinary Medicine, University of Florida, Gainesville, FL 32610, USA; [email protected] 3 College of Veterinary Medicine, University of Tennessee, Knoxville, TN 37996, USA; [email protected] * Correspondence: [email protected]; Tel.: +507-517-0740 Received: 29 May 2020; Accepted: 23 June 2020; Published: 26 June 2020 Abstract: In the 21st century, three highly pathogenic betacoronaviruses have emerged, with an alarming rate of human morbidity and case fatality. Genomic information has been widely used to understand the pathogenesis, animal origin and mode of transmission of coronaviruses in the aftermath of the 2002–2003 severe acute respiratory syndrome (SARS) and 2012 Middle East respiratory syndrome (MERS) outbreaks. Furthermore, genome sequencing and bioinformatic analysis have had an unprecedented relevance in the battle against the 2019–2020 coronavirus disease 2019 (COVID-19) pandemic, the newest and most devastating outbreak caused by a coronavirus in the history of mankind. Here, we review how genomic information has been used to tackle outbreaks caused by emerging, highly pathogenic, betacoronavirus strains, emphasizing on SARS-CoV, MERS-CoV and SARS-CoV-2. -

Respiratory Pathogen Panel
510(k) SUBSTANTIAL EQUIVALENCE DETERMINATION DECISION SUMMARY A. 510(k) Number: K152386 B. Purpose for Submission: This is a new 510(k) application for a qualitative Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR) assay used with the MAGPIX instrument for the in vitro qualitative detection of Influenza A virus (with subtype differentiation), Influenza B virus, Respiratory Syncytial virus (RSV) A and RSV B, Coronaviruses 229E, OC43, NL63 and HKU1, Human Metapneumovirus, Rhinovirus/Enterovirus, Adenovirus, Parainfluenza virus Types 1, 2, 3, and 4, Human Bocavirus, Chlamydophila pneumoniae, and Mycoplasma pneumoniae in nasopharyngeal swab (NPS) specimens from symptomatic human patients. C. Measurand: Influenza A RNA: Flu A Matrix (M) gene, Flu A H1 (HA) gene, Flu A H3 (HA) gene Influenza B RNA: Flu B Matrix (M) gene RSV A and RSV B: RNA L Polymerase gene Coronaviruses 229E, OC43 and NL63 RNA: Nucleocapsidprotein (N) gene Coronavirus HKU1: open reading frame 1 ab Human Metapneumovirus RNA: Phosphoprotein (P) gene Rhinovirus/Enterovirus RNA: 5’-UTR Adenovirus DNA: Hexon gene Parainfluenza virus RNA: Parainfluenza 1 HN gene, Parainfluenza 2 and 3 NP gene, Parainfluenza virus 4 phosphoprotein (P) gene Human Bocavirus DNA: NP1 gene Chlamydophila pneumoniae DNA: rpoB gene Mycoplasma pneumoiae DNA: P1 gene D. Type of Test: Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR) E. Applicant: Luminex Molecular Diagnostics, Inc. F. Proprietary and Established Names: NxTAG® Respiratory Pathogen Panel 1 G. Regulatory Information: -

Alkaloids: Therapeutic Potential Against Human Coronaviruses
molecules Article Alkaloids: Therapeutic Potential against Human Coronaviruses 1, 2 3 Burtram C. Fielding y , Carlos da Silva Maia Bezerra Filho , Nasser S. M. Ismail and 2, , Damião Pergentino de Sousa * y 1 Molecular Biology and Virology Research Laboratory, Department of Medical Biosciences, University of the Western Cape, Bellville 7535, South Africa; bfi[email protected] 2 Department of Pharmaceutical Sciences, Federal University of Paraíba, Paraíba 58051-900, Brazil; [email protected] 3 Pharmaceutical Chemistry Department, Faculty of Pharmaceutical Sciences and Pharmaceutical Industries, Future University in Egypt, Cairo 12311, Egypt; [email protected] * Correspondence: [email protected]; Tel.: +55-833-216-7347 These authors contributed equally to this work. y Academic Editor: Maria José U. Ferreira Received: 30 September 2020; Accepted: 10 November 2020; Published: 24 November 2020 Abstract: Alkaloids are a class of natural products known to have wide pharmacological activity and have great potential for the development of new drugs to treat a wide array of pathologies. Some alkaloids have antiviral activity and/or have been used as prototypes in the development of synthetic antiviral drugs. In this study, eleven anti-coronavirus alkaloids were identified from the scientific literature and their potential therapeutic value against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is discussed. In this study, in silico studies showed an affinity of the alkaloids for binding to the receptor-binding domain of the SARS-CoV-2 spike protein, putatively preventing it from binding to the host cell. Lastly, several mechanisms for the known anti-coronavirus activity of alkaloids were discussed, showing that the alkaloids are interesting compounds with potential use as bioactive agents against SARS-CoV-2. -

Etiology and Clinical Characteristics of SARS-Cov-2 and Other Human Coronaviruses Among Children in Zhejiang Province, China
Zhang et al. Virol J (2021) 18:89 https://doi.org/10.1186/s12985-021-01562-8 RESEARCH Open Access Etiology and clinical characteristics of SARS-CoV-2 and other human coronaviruses among children in Zhejiang Province, China 2017–2019 Yanjun Zhang1,2†, Lingxuan Su1†, Yin Chen2, Sicong Yu1, Dan Zhang3, Haiyan Mao2 and Lei Fang2* Abstract Background: A novel coronavirus (SARS-CoV-2) emerging has put global public health institutes on high alert. Little is known about the epidemiology and clinical characteristics of human coronaviruses infections in relation to infec- tions with other respiratory viruses. Methods: From February 2017 to December 2019, 3660 respiratory samples submitted to Zhejiang Children Hospi- tal with acute respiratory symptoms were tested for four human coronaviruses RNA by a novel two-tube multiplex reverse transcription polymerase chain reaction assays. Samples were also screened for the occurrence of SARS-CoV-2 by reverse transcription-PCR analysis. Results: Coronavirus RNAs were detected in 144 (3.93%) specimens: HCoV-HKU1 in 38 specimens, HCoV-NL63 in 62 specimens, HCoV-OC43 in 38 specimens and HCoV-229E in 8 specimens. Genomes for SARS-CoV-2 were absent in all specimens by RT-PCR analysis during the study period. The majority of HCoV infections occurred during fall months. No signifcant diferences in gender, sample type, year were seen across species. 37.5 to 52.6% of coronaviruses detected were in specimens testing positive for other respiratory viruses. Phylogenic analysis identifed that Zhejiang coronaviruses belong to multiple lineages of the coronaviruses circulating in other countries and areas. Conclusion: Common HCoVs may have annual peaks of circulation in fall months in the Zhejiang province, China. -
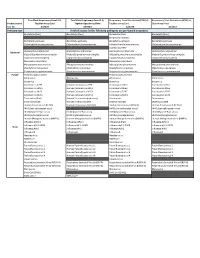
Verified Assays for the Following Pathogens Are Pre-Loaded on Product
TrueMark Respiratory Panel 2.0, TrueMark Respiratory Panel 2.0, Respiratory Tract Microbiota (RTM) v1, Respiratory Tract Microbiota (RTM) v1, Product name TaqMan Array Card TaqMan OpenArray Plate TaqMan Array Card OpenArray Plate Cat. No. A49047 A49044 A41238 A41237 Pathogen type Verified assays for the following pathogens are pre-loaded on product Bordetella (Pan) Bordetella (Pan) Bordetella (Pan) Bordetella (Pan) Bordetella holmesii Bordetella pertussis Bordetella pertussis Bordetella pertussis Bordetella pertussis Chlamydophila pneumoniae Chlamydophila pneumoniae Chlamydophila pneumoniae Chlamydophila pneumoniae Coxiella burnetii Haemophilus influenzae Haemophilus influenzae Haemophilus influenzae Haemophilus influenzae Bacterial Klebsiella pneumoniae complex Klebsiella pneumoniae complex Klebsiella pneumoniae complex Klebsiella pneumoniae complex Legionella pneumophila Legionella pneumophila Legionella pneumophila Legionella pneumophila Moraxella catarrhalis Moraxella catarrhalis Mycoplasma pneumoniae Mycoplasma pneumoniae Mycoplasma pneumoniae Mycoplasma pneumoniae Staphylococcus aureus Staphylococcus aureus Staphylococcus aureus Staphylococcus aureus Streptococcus pneumoniae Streptococcus pneumoniae Streptococcus pneumoniae Streptococcus pneumoniae Fungal Pneumocystis jirovecii Pneumocystis jirovecii Adenovirus Adenovirus Adenovirus Adenovirus Bocavirus Bocavirus Bocavirus Coronavirus 229E Human Coronavirus 229E Coronavirus HKU1 Coronavirus HKU1 Coronavirus HKU1 Human Coronavirus HKU1 Coronavirus NL63 Coronavirus NL63 Coronavirus -

Human Coronaviruses OC43 and HKU1 Bind to 9-O- Acetylated Sialic Acids Via a Conserved Receptor-Binding Site in Spike Protein Domain A
Human coronaviruses OC43 and HKU1 bind to 9-O- acetylated sialic acids via a conserved receptor-binding site in spike protein domain A Ruben J. G. Hulswita,1, Yifei Langa,1, Mark J. G. Bakkersa,1,2, Wentao Lia, Zeshi Lib, Arie Schoutenc, Bram Ophorsta, Frank J. M. van Kuppevelda, Geert-Jan Boonsb,d,e, Berend-Jan Boscha, Eric G. Huizingac, and Raoul J. de Groota,3 aVirology Division, Department of Infectious Diseases and Immunology, Faculty of Veterinary Medicine, Utrecht University, 3584 CH Utrecht, The Netherlands; bDepartment of Chemical Biology and Drug Discovery and Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CG Utrecht, The Netherlands; cCrystal and Structural Chemistry, Bijvoet Center for Biomolecular Research, Faculty of Sciences, Utrecht University, 3584 CH Utrecht, The Netherlands; dDepartment of Chemistry, University of Georgia, Athens, GA 30602; and eComplex Carbohydrate Research Center, University of Georgia, Athens, GA 30602 Edited by Mary K. Estes, Baylor College of Medicine, Houston, TX, and approved December 19, 2018 (received for review June 6, 2018) Human betacoronaviruses OC43 and HKU1 are endemic respiratory 20-nm peplomers or “spikes” that are very much a CoV hallmark pathogens and, while related, originated from independent zoo- and comprised of homotrimers of spike (S) protein, and 8-nm notic introductions. OC43 is in fact a host-range variant of the protrusions, unique to this clade, comprised of the homodimeric species Betacoronavirus-1, and more closely related to bovine coro- hemagglutinin-esterase (HE). S is central to viral entry and the — — navirus (BCoV) its presumptive ancestor and porcine hemagglu- key determinant of host and tissue tropism (11). -

Coronavirus HKU1 Infection in the United States Frank Esper,* Carla Weibel,† David Ferguson,† Marie L
Coronavirus HKU1 Infection in the United States Frank Esper,* Carla Weibel,† David Ferguson,† Marie L. Landry,† and Jeffrey S. Kahn† In 2005, a new human coronavirus, HCoV-HKU1, was The identification of the severe acute respiratory syn- identified in Hong Kong. We screened respiratory speci- drome–associated CoV in 2003 sparked renewed interest mens collected from December 16, 2001, to December 15, in the study of HCoV (7), and 4 previously unidentified 2002, from children <5 years of age who tested negative for HCoV have subsequently been discovered. HCoV-NL63, respiratory syncytial virus, parainfluenza viruses, influenza HCoV-NL, and the New Haven coronavirus (HCoV-NH) virus, and adenovirus for HCoV-HKU1 by reverse transcrip- tion–polymerase chain reaction. Overall, 1,048 respiratory are closely related group I CoV and likely represent strains specimens from 851 children were tested, and 9 HCoV- of the same species of virus (8–10). HCoV-NL63 and HKU1–positive children (1%) were identified, 2 of whom HCoV-NL were originally identified by cell culture tech- had 2 positive specimens. Children who had HCoV-HKU1 niques, while HCoV-NH was discovered by using broadly infection had evidence of either upper or lower respiratory reactive CoV molecular probes. These related viruses were tract infection or both. Two patients had disease beyond the identified in both children and adults with respiratory tract respiratory tract. HCoV-HKU1 was identified from disease. HCoV-NH was found in 8.8% of children <5 years December 2001 to February 2002. Sequence analyses of age whose specimens originally tested negative for suggest that a single strain was circulating.